Medicinal & Aromatic Plants
Open Access
ISSN: 2167-0412
![]() +44 1300 500008
+44 1300 500008
ISSN: 2167-0412
![]() +44 1300 500008
+44 1300 500008
Research Article - (2015) Volume 4, Issue 5
Withaferin A (Wi-A), a potent anticancer withanolide extracted from Withania somnifera- a tropical medicinal plant, was earlier shown to activate p53 tumor suppressor and oxidative stress pathways in cancer cells causing either growth arrest or apoptosis. In view of the information that alkyl derivatives of several bioactive steroidal compounds exhibit improved stability and activity through mechanisms that have not been completely understood, we investigated the effect of Withaferin A and its methoxy-derivative (2,3-dihydro-3- methoxy-Withaferin A) on human cancer cells to analyze the molecular interactions and efficacy. Systematic bioinformatics and molecular experimental approaches comprising expression analysis and imaging techniques were employed to demonstrate the interactions of Withaferin A and 3β-methoxy Withaferin A with their molecular targets. We demonstrate that 3β-methoxy derivation of Withaferin A possess weaker docking potential to its molecular targets and therefore possess attenuated anticancer activity as compared to Withaferin A, and was affirmed by a number of biochemical and in vitro experiments. Based on the Bioinformatics and experimental data we report that the 3β-methoxy derivation of Withaferin A attenuates its anticancer activities, and hence sufficient care is warranted in the use of these phytochemicals as anticancer reagents.
Keywords: Withania somnifera; Withaferin A; Methoxy-Withaferin A; p53; Apoptosis
Wi-A: Withaferin A; mWi-A: 3β-methoxy Withaferin A; DMSO: Dimethyl sulfoxide
Withaferin A (Wi-A) was the first member of the withanolide family to be isolated from the roots of Ashwagandha (Withania somnifera). Several others including Withanolide A, Withanolide B, Withanolide D, Withanone and their derivatives [1-4] have subsequently been identified and shown to possess bioactivities in animal as well as cell culture experimental models [5]. The systemic clinical use of this herb has not yet been possible due to the lack of identification of (i) active constituents and their biological activities, (ii) cellular targets, (iii) bioavailability and efficacy profiles of its constituent phytochemicals and (iv) pharmokinetics. Amongst various studies describing activities of Wi-A using animal and cell culture models, the anticancer activity is most supported, involving a number of pathways, identified over the years [6-12]. Furthermore, treatment with Wi-A has been suggested as a radiosensitizer in cancer treatment [13,14]. Comparison of the cytotoxicity and cytoprotective heat shock-inducing activity of Wi-A and its 36 derivatives revealed that the enone ring is an essential component for this activity. Chemical modifications, such as, acetylation or hydroxylation affected both activities suggesting that the understanding of the structure-function association may inspire development of new drugs [4]. Specifically, alkylated (e.g., methyl or ethyl) secondary metabolites have been shown to acquire greater bioactivity, metabolic stability and chemopreventive potential [15,16]. Taking this into account, we investigated the molecular interactions and potency of Withaferin A and its methoxy-derivative (2,3-dihydro- 3-methoxy-Withaferin A). In line with reported anticancer potential of Withaferin A identified in several studies [11,17,18], we investigated the efficacy of Wi-A and mWi-A as anticancer drugs by bioinformatics and biochemical experimental approaches.
Molecular docking studies
Protein preparation: The three-dimensional atomic coordinates of mortalin, p53, NRF-2 and p21WAF1 proteins (PDB-ID: 3N8E, 3D09, 2FLU and 1AXC) were downloaded from its archives the Protein Data Bank (PDB). All the crystallographic water molecules were removed from the structures, and hydrogen atoms were added using the “Hydrogen” module and correct ionization and tautomeric states were obtained. Further, Kollman united atom partial charges and salvation parameters were assigned. At the end of the protein preparation process, a PDBQT file was obtained (for each protein molecule) which was essential for the execution of AutoGrid and AutoDock.
Ligand preparation: The three-dimensional coordinates of the molecule Withaferin A (Wi-A) was used as a template to model methoxy-Withaferin A (mWi-A). The energy was minimized using GROMOS 96.1 force field through PRODRG server with default parameters.
Ligand docking: AutoDock 4.2 suite (an automated tool for the prediction of protein-ligand interactions) was used. The Lamarckian Genetic Algorithm was used with a population of 250 dockings. As the three-dimensional structures were native (not complexed with any small molecule) an appropriate grid box was generated to cover the entire protein molecule and docking was performed. Based on the results, another grid box was generated in order to cover the possible ligand-binding site and the final docking calculations were performed. Similar conformations were clustered based on the root mean square deviations (RMSD’s) and orientations.
Biochemical and visual assays
Cell culture and cytotoxicity assays: Human bone cancer (U2OS) cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, Carlsbad, CA)-supplemented with 10% fetal bovine serum in a humidified incubator (37°C and 5% CO2). For cytotoxicity assays, cells (5000/well in three independent replicates) were plated in 96-well plate and were treated with indicated doses of either Withaferin A (Wi-A) or methoxy-Withaferin A (mWi-A). Viability of control and treated cells was determined using tetrazolium dye MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT reagent, Roche), incubated for 4 h. In order to compare the long term cell proliferation and colony-forming efficiency of control and treated cells, cells (500/well in triplicates) were plated in 6-well plates and allowed to grow for 10-15 days under conditions of control or treated (as indicated). Medium was replaced every 3-4 days. Experiment was terminated by fixation of the cells in ice-cold methanol and acetone (1:1) solution for 10 min. Colonies were stained with 0.1% crystal violet. Plates were air-dried and scanned for record. Colonies were counted and statistical analysis of the colony forming efficiency in control and treated cultures was determined as described below.
Immuno-blotting: Cell pellets were solubilized in RIPA buffer (Thermo Scientific, Waltham, MA). Supernatant containing 15-20 μg of proteins was resolved in SDS-polyacrylamide gel, and was electroblotted onto PVDF membranes (Millipore, Billerica, MA) using a semidry transfer unit (ATTO, Tokyo, Japan). Immuno-blotting was performed with anti-p53, -Bcl-2, -PARP, -NRF-2, -Caspase-9 (Santa Cruz, CA); p21 (Cell Signaling) and β-actin (Abcam, Cambridge) antibodies. Anti-CARF and- mortalin antibodies were generated in our laboratory. The membranes probed with the first antibodies were excessively washed with TBS-T (Tris buffer saline-Tween 20) and incubated with secondary horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit (Santa Cruz) antibodies. Protein bands were detected using ECL prime substrate (GE Healthcare, CA). Densito-metric quantitation of three independent immuno-blotting experiments was performed with the ImageJ software (National Institute of Health). Expression level of each of the proteins in control and treated cells was calculated with respect to the β-actin (loading control). All the experiments were performed in triplicate.
Immuno-staining: Cells (4 × 104/well) were plated on the glass coverslips placed in a 12-well plate. After the cells had attached well to the surface they were treated with indicated doses of either Wi-A or mWi-A for 24 h and then fixed with either pre-chilled methanol: acetone (1:1) or 4% formaldehyde for 10 min on ice or at room temperature, respectively. Fixed cells were permeabilized with PBS-Triton-X-100 (0.1%) for 10 min followed by blocking with 2% BSA for 10 min. The coverslips were incubated with antibodies against p53, NRF-2 (Santa Cruz), p21WAF1 (Cell Signaling Technology) CARF, mortalin at room temperature (1 h) or 4oC (overnight). Cells were then probed with Alexa Fluor-conjugated secondary antibodies (Molecular Probes, Invitrogen), and counterstained with Hoechst 33258 (Sigma-Aldrich). The stained cells were viewed under Zeiss Axioplan 2 microscope and images were captured using a Zeiss AxioCam HRc camera.
Reverse-transcription PCR (RT-PCR): Total RNA was extracted with QIAGEN RNAeasy kit. Complementary DNA (cDNA) was synthesized from 2 μg of RNA using the Thermoscript Reverse Transcriptase (QIAGEN, Germany) following the manufacturer’s protocol. The synthesized cDNA was subjected to PCR amplification consisting of an initial 10 min denaturation step at 95°C followed by 30 cycles at 95°C for 45 s, 56°C for 1 min and 72°C for 45 s, with final annealing step at 72°C for 10 min. PCR amplifications were performed using specific primers for (i) p53-Sense: 5′-CTGCCCTCAACAAGATGTTTTG-3′& Antisense: 5′-CTATCTGAGCAGCGCTCATGG-3′, (ii) p21WAF1: Sense: 5′-ATGAAATTCACCCCCTTTCC-3′& Antisense: 5′-CCCTAGGCTGTGCTCACTTC-3′, (iii) CARF: Sense: 5′-CAGCCAAAGCAGCAGCAGCG-3′ & Antisense: 5′-AGCCCAACAAGGGCACCTCG-3′, (iv) NRF2: Sense: TACTCCCAGGTTGCCCACA-3′ & Antisense: 5′-CATCTACAAACGGGAATGTCTGC-3′, (v) GAPDH: Sense: 5’-ACCTGACCTGCCGTCTAGAA-3’ & Antisense: 5’-TCCACCACCCTGTTGCTGTA-3’. The PCR products were then run on a 1% agarose gel and stained with ethidium bromide for visualization.
Cell cycle analysis: Assessment of different phases of cell cycle was performed through PI cell cycle staining. Cells were harvested (Trypsin- EDTA) and fixed in 70% chilled ethanol for 15-30 min at 4°C. Cell pellets was suspended in PBS and incubated with 50 μl Ribonuclease A (5 mg/ml; Qiagen) for 30 min at 37°C to degrade cellular RNA content. Cells were stained with 250 μl Guava cell cycle reagent (50 μg/ml) for 1 h in dark and analyzed on the Guava cell cycle analyzer (Merk, Millipore). The data were analyzed by ModFit software to evaluate the distribution of cells in different phases of cell cycle.
Statistical analysis
All experiments were carried out in triplicate. Data values were expressed as mean ± SEM of three individual experiment sets. Statistical analyses were carried out using Student’s t-test or nonparametric Mann- Whitney U-test; whichever was applicable. Statistical significance was defined as p-value ≤ 0.05.
Docking of methoxy-withaferin a with mortalin, p53, p21WAF1 and NRF-2
Withanolides, Withaferin A (Wi-A) and Withanone (Wi-N) from Ashwagandha have been shown to possess anticancer activity [12,17-19]. Whereas Wi-N killed cancer cells selectively, Wi-A was found to have cytotoxicity to normal cells also [6,19,20]. Studies on the molecular mechanism of their action have revealed that these evoke oxidative stress and DNA damage response in cancer cells, and trigger either growth arrest or apoptosis in a dose dependent manner. Loss-offunction screenings earlier revealed the involvement of mortalin, p53, p21, TPX-2, aurora, vimentin and NRF-2 proteins in Wi-A and Wi-N mediated cancer cell killing [6,19,20].
Mortalin is a mitochondrial chaperone that has been shown to be frequently up regulated in many cancers and is known to promote carcinogenesis by inactivation of the tumor suppressor protein p53 and deregulation of apoptosis. Activation of p53 by abrogation of its complex with mortalin has been shown to cause growth arrest of cancer cells in several studies [21-23]. We have earlier shown that Wi-N and Wi-A can interfere with the binding of mortalin to p53. In the present study, the modelled three-dimensional structure of 3β-methoxy-Withaferin-A (mWi-A) was docked with the substrate binding domain of mortalin (PDB-id: 3N8E) (Figure 1). As shown in Table 1, compared to Wi-A, mWi-A was less efficient both in terms of binding energy and number of hydrogen bonding interactions. Wi-A and mWi-A exhibited a binding energy of -9.8 and -7.47 Kcal/mol, respectively. Of note, both withanolides (Figure 1A) interacted with the residue 513 (Arg) of mortalin (Figure 1B). The latter constitute the “latch region” of mortalin and functions as a “latch” between the “lid and cleft regions” that are important for its chaperoning activity (Figure 1C). Thus, it was predicted that mWi-A, although weaker than Wi-A, might cause an antagonistic effect on the activity of the protein. We next examined if it would also bind to p53. The modelled structure of mWi-A was docked with the three-dimensional structure of p53 (PDB-id: 3D09). The results (Figure 1D-E and Table 1) suggested that mWi-A is less efficient (binding energy: -6.74 Kcal/mol) as compared to Wi-A (binding energy: -10.18 Kcal/mol). Similar to Wi-A, it was predicted to interact with Leu 111 and Arg 282 (Figure 1E), the residues known to be essential for the stability of the p53 protein. Furthermore, similar to Wi-A, it was predicted to disrupt the mortalin-p53 complex. Based on these analyses, it could be perceived that mWi-A may affect mortalin-p53 complex causing its activation and upregulation of p21WAF1 protein, a crucial downstream effector of p53 involved in growth arrest and initiation of S phase of the cell cycle.
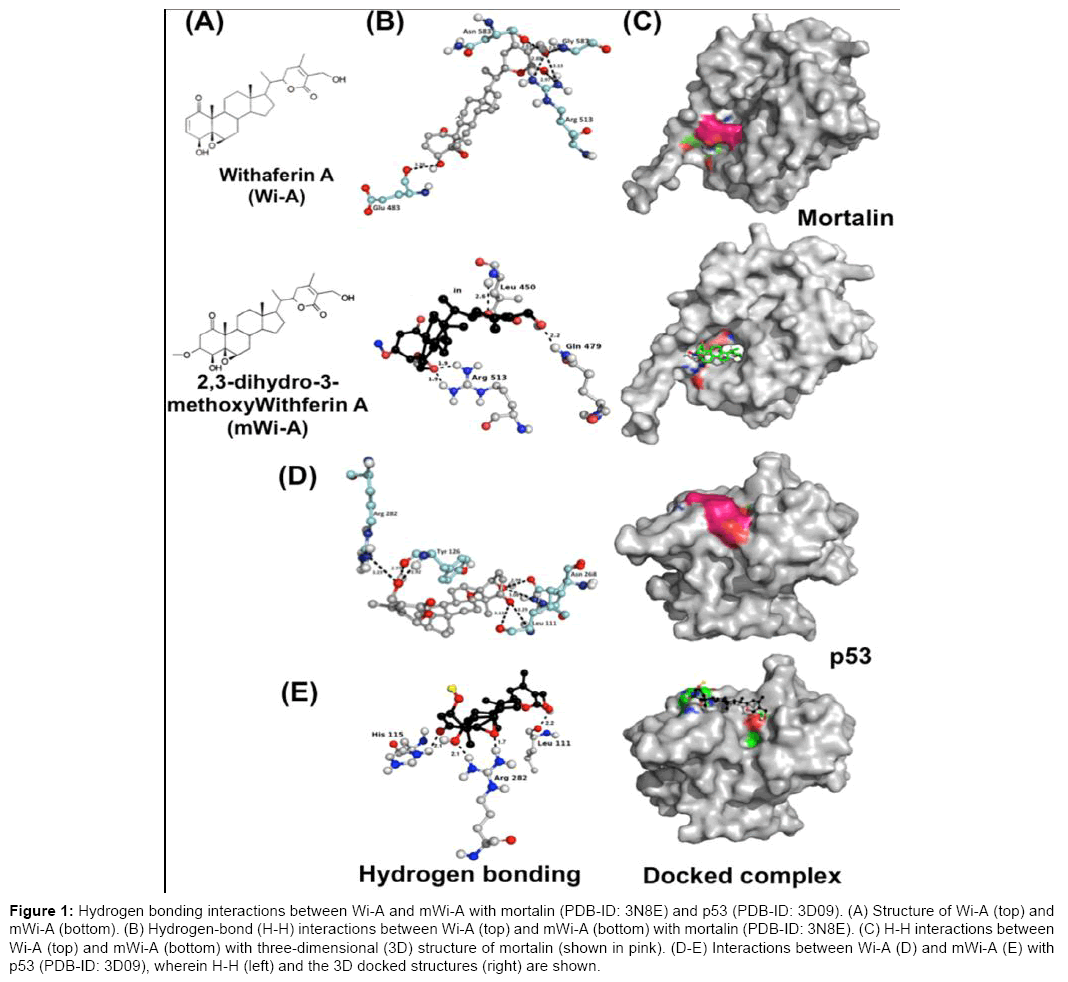
Figure 1: Hydrogen bonding interactions between Wi-A and mWi-A with mortalin (PDB-ID: 3N8E) and p53 (PDB-ID: 3D09). (A) Structure of Wi-A (top) and mWi-A (bottom). (B) Hydrogen-bond (H-H) interactions between Wi-A (top) and mWi-A (bottom) with mortalin (PDB-ID: 3N8E). (C) H-H interactions between Wi-A (top) and mWi-A (bottom) with three-dimensional (3D) structure of mortalin (shown in pink). (D-E) Interactions between Wi-A (D) and mWi-A (E) with p53 (PDB-ID: 3D09), wherein H-H (left) and the 3D docked structures (right) are shown.
| Receptor | Ligand | Hydrogen bond interactions | Distance (Å) | Binding energy Kcal/mol |
|---|---|---|---|---|
| Mortalin (PDB-ID: 3N8E) |
Wi-A | Glu (483) O2-H...O Arg (513) NH1-H…O4 Arg (513) NH1-H…O6 Arg (513) NH2-H…O6 Gly (587) N-H…O6 Asn (583) O6-H…O |
3.20 2.97 3.13 2.88 2.84 2.61 |
-9.80 |
| mWi-A | Arg(513) NH1-H...O11 Arg(513) NH2-H...O11 Leu (450) N-H...O4 Gln(479) NE2-H...O6 |
1.9 1.9 2.6 2.2 |
-7.47 | |
| p53 (PDB-ID: 3D09) |
Wi-A | Arg (282) NH1-H…O6 Tyr (126) O6-H…O Tyr (126) N-H…O6 Leu (111) O-H…O1 Leu (111) N-H…O1 Asn (268) ND2 (A)-H…O2 Asn (268) O2-H…OD1(A) |
3.23 2.77 2.92 3.13 3.29 3.08 2.98 |
-10.18 |
| mWi-A | Leu(111) O8-H...O His(115) ND1-H...O50 Arg(282) NH1-H...O5 Arg(282) NH2-H...O6 |
2.2 2.1 2.1 1.7 |
-6.74 | |
| p21WAF1 (PDB-ID: 1AXC) |
Wi-A | Met (147) O2-H…O Tyr (151) N-H…O2 His (152) O-H…O1 Ile (158) N-H…O6 Arg (156) O6-H…O Arg (156) O-H…O4 Arg (156) N-H…O5 Arg (155) NH1-H…O4 |
2.44 3.37 2.67 3.40 2.81 2.79 3.07 2.71 |
-7.84 |
| mWi-A | Arg (155) N-H...O74 Arg (156) NE-H...O4 Leu (157) O8-H...O Phe (159) N-H...O8 |
2.00 2.10 1.80 2.30 |
-6.58 | |
| NRF-2 (PDB-ID: 2FLU) |
Wi-A | Ala (69) N-H…O2 Phe (71) N-H…O2 Ala (72) N-H…O3 Gln (75) OE1-H…O4 Leu (76) O6-H…O |
2.74 3.44 2.52 3.51 2.87 |
-7.87 |
| mWi-A | Ala (69) N-H...O11 Ala (69) N-H...O8 Phe (70) N-H...O8 Phe (71) N-H...O11 |
1.8 2.3 1.9 1.8 |
-4.60 |
Table 1: Details of molecular interaction of Wi-A and mWi-A with their targets proteins.
We, thus, also investigated the docking efficacy of Wi-A and mWi-A to p21WAF1. As shown in Figure 2 and Table 1, both Wi-A and mWi-A showed interaction to the same sites of p21WAF1 (Figure 2A- 2B). However, significant difference in the binding energies of Wi-A (7.84 Kcal/mol) and mWi-A (-6.58 Kcal/mol) was observed (Table 1). Docking analysis of Wi-A and mWi-A to the three-dimensional structures of NRF-2 (PDB-id: 2FLU), an important regulator of cellular oxidative stress response revealed that they dock to the same sites (Figure 2C-2D and Table 1) and similar to the other targets (as described above) mWi-A showed weaker interaction (binding energy- 4.60 Kcal/mol) than Wi-A (binding energy -7.87 Kcal/mol). Taken together, these predictions envisaged that in contrast to Wi-A, mWi-A exhibit weaker interactions with the target proteins and hence may possess weak potency, and low efficacy as an anticancer drug.
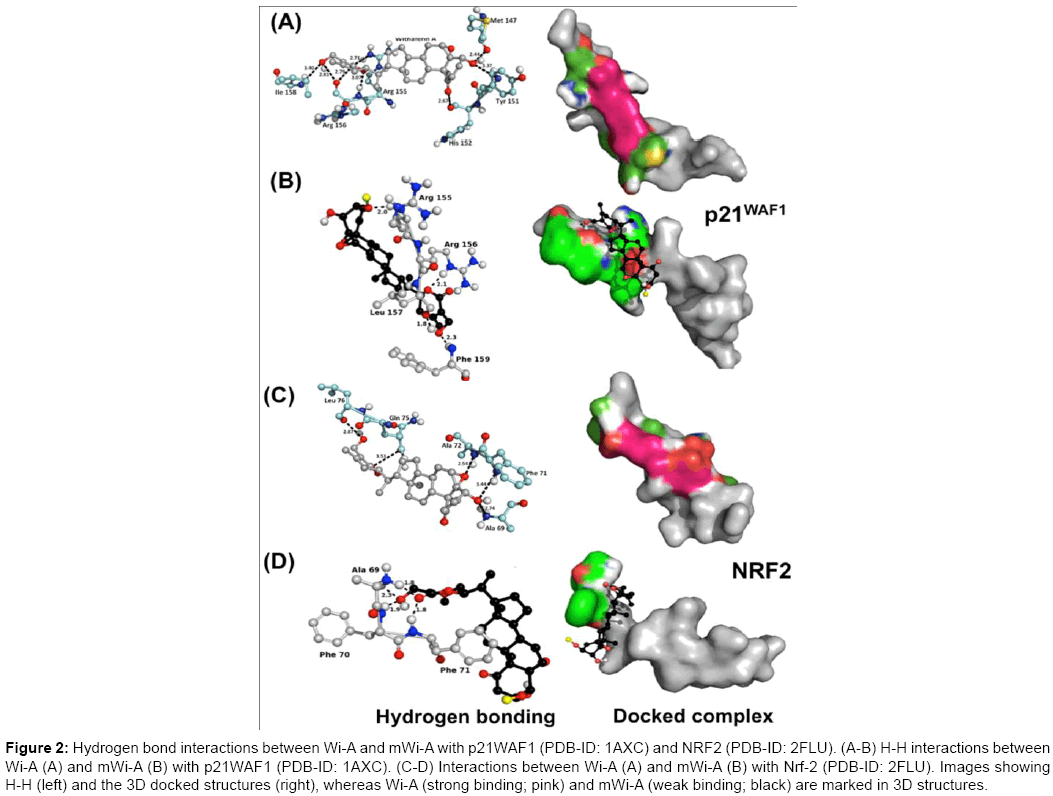
Figure 2: Hydrogen bond interactions between Wi-A and mWi-A with p21WAF1 (PDB-ID: 1AXC) and NRF2 (PDB-ID: 2FLU). (A-B) H-H interactions between Wi-A (A) and mWi-A (B) with p21WAF1 (PDB-ID: 1AXC). (C-D) Interactions between Wi-A (A) and mWi-A (B) with Nrf-2 (PDB-ID: 2FLU). Images showing H-H (left) and the 3D docked structures (right), whereas Wi-A (strong binding; pink) and mWi-A (weak binding; black) are marked in 3D structures.
Effect of Wi-A and mWi-A on p53-p21WAF1 pathway
In order to validate the above bioinformatics predictions, we examined the activation of p53 in control and treated human bone cancer (U2OS; possess wild type p53 protein) cells. Induction of p53 was observed in cells treated with Wi-A (Figure 3A), at a dose as low as 0.6 μg/ml. Whereas, mWi-A treated cells showed no change in the expression level of p53 even with five fold higher doses (3 μg/ ml) (Figure 3A). Immuno-cytochemical analysis of p53 in control and Wi-A treated cells revealed strong nuclear p53 staining in majority of the cells with doses 0.6 μg/ml and above (Figure 3B). In view of several other studies [24,25], the present data suggested the (i) transcriptional activation function of p53 and (ii) abrogation of its complex with mortalin in the cell cytoplasm. Of note, mWi-A treated cells showed no change in the staining intensity or localization of p53 (Figure 3B). We next examined the activation of p53 function by analysing the level of p21WAF1. As anticipated, p21WAF1 exhibited upregulation, affirmed in both experiments (Figure 3A and 3B), in Wi-A treated cells only. In order to delineate the mechanism of upregulation, we also performed RT-PCR analysis and found a low, but significant, upregulation of both p53 and p21WAF1 levels in only Wi-A treated cells (Supp. Figure 1).
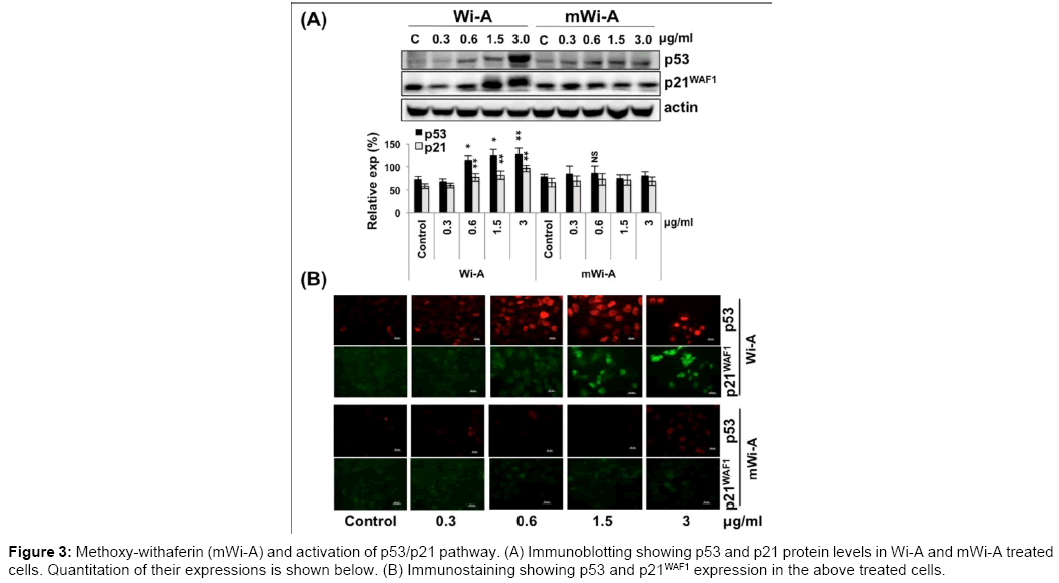
Figure 3: Methoxy-withaferin (mWi-A) and activation of p53/p21 pathway. (A) Immunoblotting showing p53 and p21 protein levels in Wi-A and mWi-A treated cells. Quantitation of their expressions is shown below. (B) Immunostaining showing p53 and p21WAF1 expression in the above treated cells.
Based on the above data, we investigated if the nuclear enrichment and activation of p53 was a result of disruption of mortalin-p53 complexes in cell cytoplasm due to strong docking of Wi-A in these proteins as predicted by bioinformatics analysis described above. Immuno-blotting and immuno-staining of mortalin in Wi-A treated and control cells showed decrease in mortalin and shift in its staining pattern from the perinuclear to pancytoplasmic region (Figure 4A and 4B; data not shown). Of note, mWi-A cells showed no difference. Consistent with this, increase in nuclear p53 was observed in Wi-A, but not in mWi-A, treated cells (Supp. Figure 2). These data endorsed the above findings that docking of Wi-A to mortalin and p53 proteins disrupted their interactions, causing nuclear translocation and activation of p53 as determined by upregulation of p21WAF1 protein. Importantly, mWi-A is found incapable to induce such activation of p53-p21 pathway.
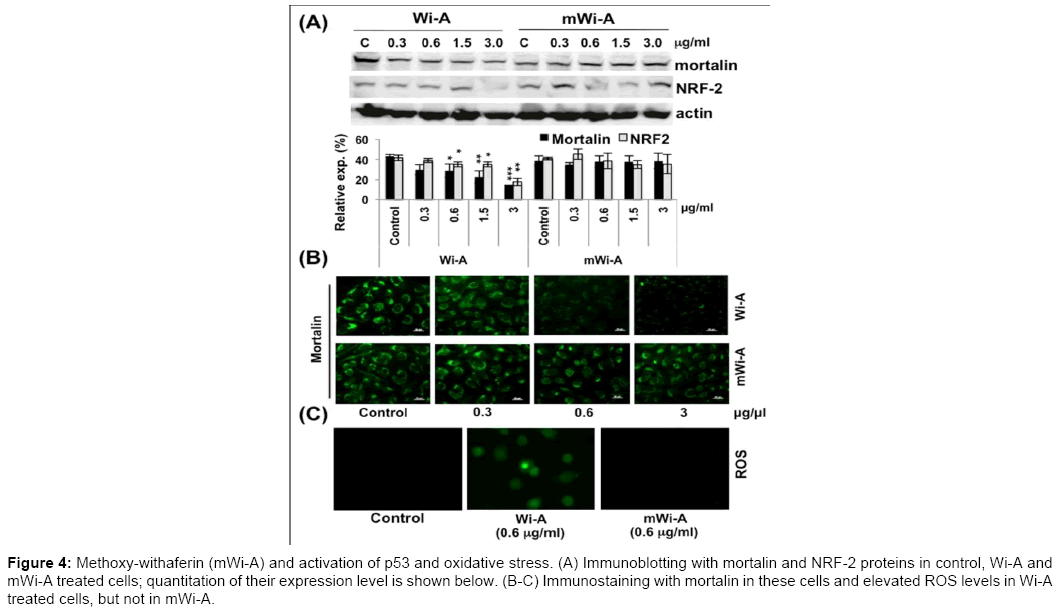
Figure 4: Methoxy-withaferin (mWi-A) and activation of p53 and oxidative stress. (A) Immunoblotting with mortalin and NRF-2 proteins in control, Wi-A and mWi-A treated cells; quantitation of their expression level is shown below. (B-C) Immunostaining with mortalin in these cells and elevated ROS levels in Wi-A treated cells, but not in mWi-A.
Induction of oxidative stress and DNA damage by Wi-A and mWi-A
NRF-2 is an important regulator of cellular oxidative stress response and shown to induce antioxidant response in cells. NRF-2 is upregulated in many cancers and provides an advantage of survival against oxidative stress. We earlier showed that NRF-2 is an important target of Wi-A [25]. The level of NRF-2 was examined in mWi-A treated and control cells. Whereas no change, either in expression level (Figure 4A) or staining intensity (Supp. Figure 3) was observed in mWi-A treated cells, high doses of Wi-A (0.6 to 3.0 μg/ml) lead to decrease in NRF-2 expression, also observed at the transcript level (Supp. Figure 4).
Consistent with these data, we found that the cells treated with Wi-A showed increase in ROS level; mWi-A treated cells remained unaffected (Figure 4C). DNA damage response has been implicated as a predominant mechanism of action of several anticancer drugs. We have earlier shown that CARF (Collaborator of ARF) is involved in DNA damage response and checkpoint signaling pathways [26]. Increase in CARF expression was seen to correlate with the increased expression of DNA damage response and checkpoint proteins. We examined the level of CARF in control, Wi-A and mWi-A treated cells. Consistent with the anticancer potency of Wi-A, we found that it resulted in moderate, but significant, induction of CARF both at the protein and mRNA levels (Figure 5A and 5B). Immunostaining of CARF revealed its upregulation in Wi-A treated cells, while its expression remain unchanged in mWi-A treated cells (Figure 5C). Therefore, mWi-A treated cells did not show such changes correlating with its low efficacy or lack of anticancer activity. Consistent with the above finding, levels of Gadd45a and γH2AX were also found upregulated in Wi-A treated cells only (data not shown).
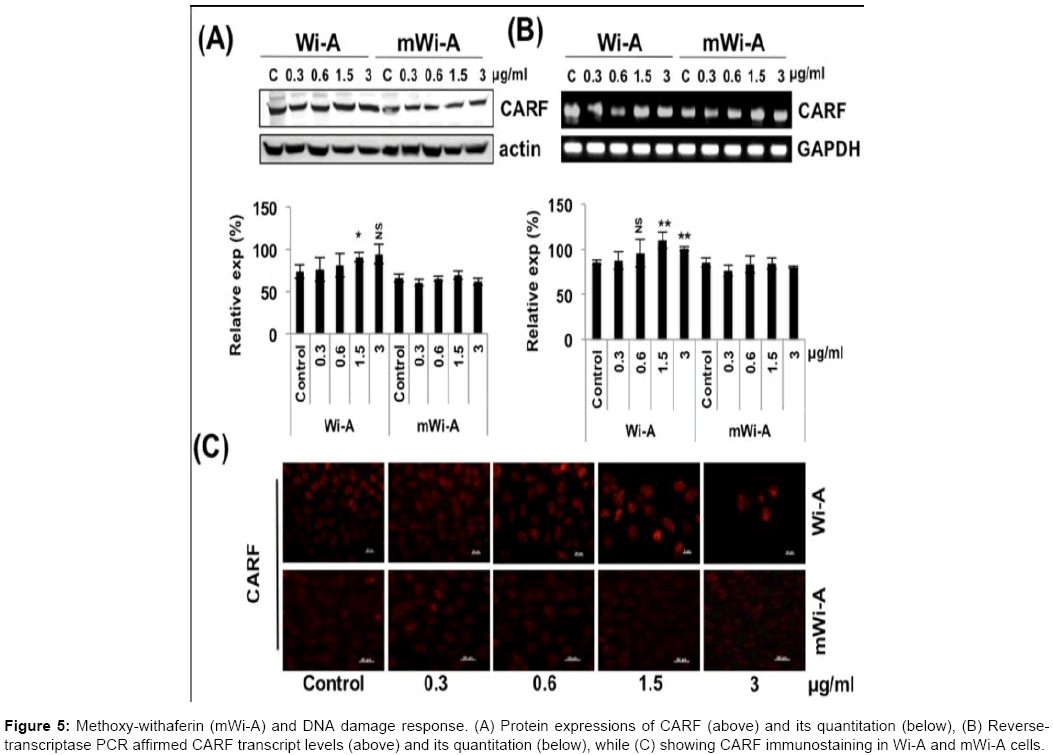
Figure 5: Methoxy-withaferin (mWi-A) and DNA damage response. (A) Protein expressions of CARF (above) and its quantitation (below), (B) Reversetranscriptase PCR affirmed CARF transcript levels (above) and its quantitation (below), while (C) showing CARF immunostaining in Wi-A and mWi-A cells.
Anticancer efficacy of Wi-A and mWi-A
We further examined the cytotoxicity of Wi-A and mWi-A by cell viability and colony forming assays. Wi-A exhibited strong cytotoxicity (Figure 6A and Supp. Figure 5). In above observation, it was found that the cells undergo growth arrest at low dose, but apoptosis at high dose of Wi-A, respectively. By contract, equivalent doses of mWi-A remained ineffective both in short-term viability and long-term clonogenic assays (Figure 6B, Supp. Figures 6 and 7). mWi-A treated cells exhibited normal morphology (Figure 6A) and showed negligible effect on the colony forming efficacy at doses as high as 3 μg/ml (Figure 6B). Cell cycle analysis also revealed G1 and G2 arrest in Wi-A, and not mWi-A, treated cells at dose of 0.3 μg/ml (Figure 6C). Whereas high doses (1.5 and 3.0 μg/ml) of Wi-A, and not mWi-A, caused apoptosis, as also supported by expression analysis of control and treated cells. Consistent with their phenotypes, down-regulation of anti-apoptotic proteins, Bcl-2, total PARP and Caspase 9 was detected in Wi-A treated cells indicating activation of apoptosis pathway in these cells, while mWi-A failed to modulate expression of apoptosis markers (Figure 6D).
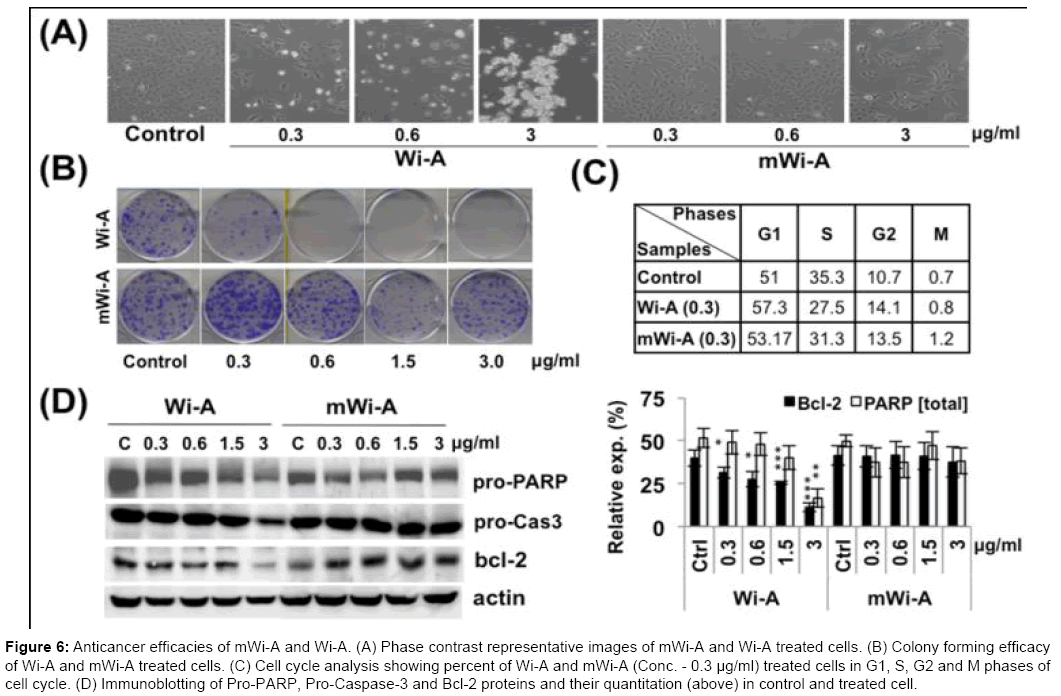
Figure 6: Anticancer efficacies of mWi-A and Wi-A. (A) Phase contrast representative images of mWi-A and Wi-A treated cells. (B) Colony forming efficacy of Wi-A and mWi-A treated cells. (C) Cell cycle analysis showing percent of Wi-A and mWi-A (Conc. - 0.3 μg/ml) treated cells in G1, S, G2 and M phases of cell cycle. (D) Immunoblotting of Pro-PARP, Pro-Caspase-3 and Bcl-2 proteins and their quantitation (above) in control and treated cell.
Advancement of computational biology has provided a comprehensive platform to model, predict and validate putative molecular interaction. It has provided an insight into detailed structural annotation that may predict atomics bonding, localization, and its strength to further affirm it in wet lab. Prediction of binding of mortalin, p53, p21WAF1 and NRF-2 with Wi-A and its methoxy derivative (mWi-A) at the first step demonstrated strength of their interactions that was further affirmed by molecular validations. Strength of mWi-A interaction with their target proteins was lower in comparison to Wi-A, and thus it reflected in further expression validations. The strong interaction of Wi-A with target proteins led to activation of p53/p21WAF1 pathway, while it modulated mortalin and NRF-2 levels. On the other hand, mWi-A remained ineffective in doing so, reflecting its weaker interaction with target proteins predicted with bioinformatics analysis. Consistent with initial predictions, Wi-A mediated activation of p53/p21 pathway further found to lead growth arrest followed by apoptosis, validated by associated markers. Altered cell cycle profiles showed increased G0/G1 and arrested S phase in Wi-A treated cells affirmed efficacy of Wi-A, and not of mWi-A. Activation of ROS and CARF expression further demonstrated induced oxidative stress and DNA damage response pathway in Wi-A treated cells, though mWi-A failed in activating above response. In conjunction with the bioinformatics analyses, our study showed that small chemical modifications i.e., methylation of withaferin A exerts major impact on its binding efficacy to the target proteins and its cellular outcomes. Moreover, alkyl modification, besides improving drug potency by stability and activity of organic/steroidal compounds, may also cause attenuation of their chemotherapeutic potency. Altogether, present investigation implies that (i) the bioinformatics predictions are reliable and good predictive parameters and (ii) sufficient care is warranted in the use of these phytochemicals as anticancer reagents.
The authors have no conflict of interest
KS, ZZ, SCK and RW designed the study and drafted the manuscript. CC, KV and RSK have carried out all the experiments. All the authors read and approved the final version of the manuscript.
The study was supported by grants from the AIST. RSK is a recipient of Japan Society for Promotion of Science (JSPS) Post-doctoral Fellowship. Authors thank Navjot Shah, Anupama Chaudhary and Jay Prakash for their contributions to this study.