Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2015) Volume 2, Issue 2
A three dimensional quantitative structure activity relationship (3D QSAR) using k nearest neighbor molecular field analysis (kNN MFA) method was performed on a series of isatin derivatives as carboxylesterase (CE) inhibitors. This study was performed with 49 compounds (data set) using sphere exclusion (SE) algorithm for the division of the data set into training and test set. SE algorithm allows constructing training sets covering all descriptor space areas occupied by representative points. Between 3.0 to 5.5 dissimilarity levels which comprises of test set size 4 to 10, kNN-MFA methodology with stepwise (SW), simulated annealing (SA) and genetic algorithm (GA) was used for building the QSAR models. Four predictive models were generated with SW-kNN MFA (pred_r2=0.7552 to 0.9376), three predictive models were generated with SA-kNN MFA (pred_r2=0.7019 to 0.9367) and two predictive models were generated with GA-kNN MFA (pred_r2=0.8226 to 0.8497). Most significant model generated by stepwise kNNMFA showed internal predictivity 82.11% (q2=0.8211) and external predictivity 93.76% (q2=0.9376). In this model hydrophobic and steric interactions dominate the CE inhibitory activity. Hydrophobic field descriptor (H_977) with positive range indicates that positive hydrophobic potential is favorable for increase in activity and hence more hydrophobic substituent group is preferred in that region. Steric field descriptor (S_619) with negative range indicates that negative steric potential is favorable for increase in activity and hence less bulky substituent group is preferred in that region. The kNN-MFA contour plots provided further understanding of the relationship between structural features of substituted isatin derivatives and their activities which should be applicable to design newer potential CE inhibitors.
<Keywords: 3D-QSAR; kNN-MFA; Carboxylesterases inhibitors; Isatin derivatives
Carboxylesterases (CE) are ubiquitous enzymes which are responsible for the metabolism and detoxification of xenobiotics. Carboxylesterases (CEs) hydrolyze endogenous and exogenous esters to the corresponding alcohol and carboxylic acid [1-4]. In mammals, they tend to be expressed in tissues likely to be exposed to xenobiotics, including the liver, lung, small intestine, kidney, and so on [5]. CEs metabolize a number of useful drugs [6-9] such as Demerol and lidocaine, the anticancer agents capecitabine and CPT-11 [10] (irinotecan, 7-ethyl-10-[4-(1-piperidino)-1-piperidino] carbonyloxycamptothecin), breast cancer drug tamoxifen [11], antiviral drug oseltamivir, alzheimer’s drug tacrine [12], narcotics cocaine and heroin [13], antithrombogenic agent clopidogrel, cholesterol lowering drug mevastatin [11]. Therefore, identification and application of selective CE inhibitors may prove useful in modulating the metabolism of esterified drugs in-vivo and for prolonging the bioactivity of agents that are inactivated by CEs or conversely may reduce the toxicity of compounds that are activated by these enzymes.
Benzil, ethane-1,2-dione chemo type (Benzil based compounds [14-17], analogues of isatin [18] reported in literature as CE inhibitors. Quantitative structure-activity relationships (QSAR) represent an attempt to correlate properties (descriptors) of compounds with activities. The QSAR relationship is expressed as a mathematical equation [19-23]. Isatin analogs [18] with varied mode of selectively inhibits the Carboxylesterases (CE) were selected and 3D QSAR studies were performed to generate best predicative and validated QSAR model. In the present study, we have applied k-nearest neighbor molecular field analysis (kNNMFA) [24,25]. 3D-QSAR models permitted an understanding of the steric, electrostatic and hydrophobic requirements for ligand binding.
Data set and biological activity
¨In the present study 49 molecules of Isatin (indole-2,3-dione) derivatives [18] were used which were reported to have carboxylesterase inhibitory activity. Human intestinal carboxylesterase (hiCE) inhibitory data Ki [inhibition constant (μM)] reported have been converted to the logarithmic scale [pKi (moles)] for QSAR study.
Molecular modeling study
Molecular modeling and kNN-MFA studies were performed on HCL computer having genuine Intel Pentium Dual Core Processor and Windows XP operating system using the software Molecular Design Suite (MDS) [26]. Structures were drawn using the 2D draw application and converted to 3D structures. Structures were optimized by energy minimization and geometry optimization was done using Merck Molecular Force Field (MMFF) method with 10000 as maximum number of cycles, 0.01 as convergence criteria (root mean square gradient) and 1.0 as constant (medium’s dielectric constant which is 1 for in vacuo) in dielectric properties. The default values of 30.0 and 10.0 Kcal/mol were used for electrostatic and steric energy cutoff.
Molecular alignment: The dataset were aligned by template based alignment method using most active molecule (49, Table 1) as a reference molecule 2 and structure 1 as a template (Figure 1) for alignment of the molecules. The alignment of all the molecules on the template (Figure 2). In the template based alignment method, a template structure was defined and used as a basis for alignment of a set of molecules.
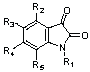
| S.No. | ID | R1 | R2 | R3 | R4 | R5 | Ki (hiCE) [mM] | log (1/Ki) or pKi [M] |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | CH3 | 38.2 | 4.417 | ||||
| 2 | 3 | CH2OH | 34.5 | 4.4621 | ||||
| 3 | 4 | Cl | 29.2 | 4.5346 | ||||
| 4 | 5 | C2H4Br | 1.58 | 5.8013 | ||||
| 5 | 6 | C2H4l | 0.74 | 6.1307 | ||||
| 6 | 10 | COC3H7 | 68.2 | 4.1662 | ||||
| 7 | 13 |  |
0.95 | 6.0222 | ||||
| 8 | 14 | 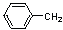 |
0.87 | 6.0604 | ||||
| 9 | 15 |  |
0.032 | 7.4948 | ||||
| 10 | 16 | 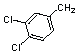 |
0.067 | 7.1739 | ||||
| 11 | 17 | 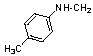 |
1.08 | 5.9665 | ||||
| 12 | 18 | 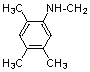 |
2.88 | 5.5406 | ||||
| 13 | 19 | 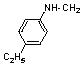 |
0.41 | 6.3872 | ||||
| 14 | 20 | 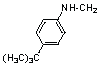 |
0.61 | 6.2146 | ||||
| 15 | 21 | 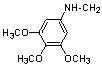 |
2.69 | 5.5702 | ||||
| 16 | 22 | 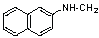 |
0.11 | 6.9586 | ||||
| 17 | 23 | 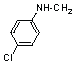 |
0.2 | 6.6989 | ||||
| 18 | 24 | 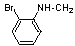 |
0.047 | 7.3279 | ||||
| 19 | 25 |  |
0.17 | 5.7695 | ||||
| 20 | 27 | 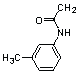 |
5.51 | 5.2588 | ||||
| 21 | 28 | C2H5 | 37.9 | 4.4213 | ||||
| 22 | 29 | Cl | 7.77 | 5.1095 | ||||
| 23 | 33 | Cl | 14.9 | 4.8268 | ||||
| 24 | 34 | Br | 13.3 | 4.8761 | ||||
| 25 | 35 | 22.8 | 4.6420 | |||||
| 26 | 36 | CF3O | 7.49 | 5.0990 | ||||
| 27 | 38 | CH2(C=CH2)CH3Cl | Br | 0.28 | 6.5528 | |||
| 28 | 40 | Cl | 29.7 | 4.5361 | ||||
| 29 | 43 | Cl | 9.55 | 5.0199 | ||||
| 30 | 45 | CF3 | 33.4 | 4.4762 | ||||
| 31 | 47 | CH3 | CH3 | 30.8 | 4.5114 | |||
| 32 | 48 | CH3 | CH3 | 84.6 | 4.0736 | |||
| 33 | 49 | Cl | Cl | 2.56 | 5.5917 | |||
| 34 | 50 | Cl | Cl | 0.62 | 6.2076 | |||
| 35 | 51 | Cl | 0.65 | 6.1870 | ||||
| 36 | 52 | Cl | Cl | 16.6 | 4.7798 | |||
| 37 | 53 | Cl | Cl | 4.08 | 5.3893 | |||
| 38 | 54 | Cl | Cl | 21.8 | 4.6615 | |||
| 39 | 55 | Cl | CH3 | 17.2 | 4.7520 | |||
| 40 | 56 | Cl | CH3 | 21.8 | 4.6615 | |||
| 41 | 57 | Cl | CH3 | 53.5 | 4.2716 | |||
| 42 | 58 | CH3 | Br | 7.41 | 5.1301 | |||
| 43 | 59 | 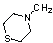 |
22.2 | 4.6536 | ||||
| 44 | 60 |  |
27.4 | 4.5622 | ||||
| 45 | 61 | 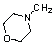 |
22.5 | 4.478 | ||||
| 46 | 62 | c12h25c | 0.008 | 8.0969 | ||||
| 47 | 63 | C16H33C | 0.011 | 7.9586 | ||||
| 48 | 64 |  |
1.54 | 5.8124 | ||||
| 49 | 65 | 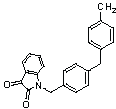 |
0.01 | 8.0000 |
Table 1: General structure of Isatin derivatives and their biological activities (data set of 49 molecules).
*Compounds having Ki>100 (µM) in the selected series were excluded and the resulting total data set of 49 molecules for hiCE were used for the present study.

Figure 1: Structure of template [1] and reference molecule [2] used in template based alignment.

Figure 2: 3D-alignment of molecules (template based).
Grid size: Once the molecules are aligned, a grid or lattice is established which surrounds the set of compounds in potential receptor surface. Current study uses grid resolution 2 Å.
Descriptor calculation: Once the molecules are aligned, a molecular field is computed on a grid of points in space around the molecule. This field provides a description of how each molecule will tend to bind in the active site. Descriptors representing the steric, electrostatic and hydrophobic interaction energies were computed at the lattice points of the grid using a methyl probe of charge +1.
Generation of training and test set: Sphere exclusion algorithm was used for generation of training and test sets. The whole data set was divided into training and test sets using sphere exclusion algorithm [25]. This algorithm allows constructing training sets covering all descriptor space areas occupied by representative points. The higher the dissimilarity level c is, smaller the training set and larger the test set and vice versa. It is expected that the predictive ability of QSAR models generally decrease when the dissimilarity level increases. Once the training and test sets are generated, kNN methodology is applied to descriptors generated over grid.
kNN-MFA methodology for building QSAR models: Models generated by kNN-MFA in conjunction with stepwise (SW) forwardbackward, simulated annealing (SA) and genetic algorithm (GA) variable selection methods. The QSAR models were developed using Stepwise forward-backward variable selection method with pKi activity field as dependent variable and descriptors as independent variable.
The kNN technique is a conceptually simple approach to pattern recognition problems. In this method, an unknown pattern is classified according to the majority of the class memberships of its k nearest neighbors in the training set. The nearness is measured by an appropriate distance metric (e.g., a molecular similarity measure, calculated using field interactions of molecular structures).
Stepwise forward-backward variable selection method [27] (SWkNN MFA): This method uses stepwise variable selection and k-NN principle to build QSAR model. In each step this method optimize (i) the number of nearest neighbors (k) used to estimate the activity of each compound and (ii) select variables (stepwise) from the original pool of all molecular descriptors that are used to calculate similarities between compounds. It involves a step-by-step search procedure that begins by addition of a single independent variable with optimal k value (optimizing k value from the given range of k values) and highest sum of weighted k-nearest neighbor cross validation (q2) and external validation (pred_r2) value amongst all available descriptors to form a model.
The parameter settings used for SW-kNN MFA are Cross correlation limit as 0.5, maximum number of variable in final equation as n/5 (n is number of compounds in training set), term selection criteria as q2, Ftest in as 4 and Ftest out as 3.99, variance cut-off as 0 and Scaling as Auto Scaling, number of maximum neighbors as 5, number of minimum neighbors as 2 and distance based weighted average as prediction method.
Simulated annealing k-NN QSAR algorithm [24] (SA-kNN MFA): Simulated annealing is to simulate a physical process called annealing, in which a system is heated to a high temperature and then is gradually lowered to a preset temperature value (e.g. room temperature). During this process, the system samples possible configurations according to Boltzmann distribution. At equilibrium, low energy states wil be mostly populated.
k-NN MFA method employs the kNN classification principle combined with the variable selection procedure. For each predefined number of variables (nvar) it seeks to optimize using stochastic sampling and simulated annealing as an optimization tool. The parameter settings used for simulated annealing in the present study are Maximum temperature as 100, minimum temperature as 0.01, cross correlation limit as 0.5, terms in model as n/5 (n is number of compounds in training set), iteration at given temperature as 5, decrease temperature by as 10, seed as 0, perturbation limit as 1, term selection criteria as q2, variance cut-off as 0 and Scaling as Auto Scaling, number of maximum neighbors as 5, number of minimum neighbors as 2 and distance based weighted average as prediction method.
Genetic algorithm k-NN QSAR algorithm (GA-kNN MFA): The genetic function approximation (GFA) algorithm (Rogers and Hopfinger) offers a new approach in building quantitative structureactivity relationship (QSAR) and quantitative structure-property relationship (QSPR) models. Replacing regression analysis with the GFA algorithm allows the construction of models competitive with or superior to those produced by standard techniques and makes available additional information not provided by other techniques [28].
The GFA algorithm used to perform a search over the space of possible QSAR/QSPR models using the fitness score (LOO q2/cross terms) to estimate the fitness of each model. Such evolution of a population of randomly constructed models leads to the discovery of highly predictive QSARs/QSPRs.
The parameter settings used for genetic algorithm in the present study are Cross correlation limit as 0.5, chromosome length as n/5 (n is number of compounds in training set), cross over probability as 0.95, mutation probability as 0.05, population as 10, number of generations as 1000, convergence criteria as 0.01, seed as 0, term selection criteria as q2, variance cut-off as 0 and Scaling as Auto Scaling, number of maximum neighbors as 5, number of minimum neighbors as 2 and distance based weighted average as prediction method.
Cross-validation (q2) using weighted k-nearest neighbor [25]: In cross validation, a compound is eliminated in the training set and its biological activity is predicted on the basis of the k-NN principle, i.e., as the weighted average activity of k most similar molecules (k is set to 1 initially). The similarities are evaluated as Euclidean distances between compounds using only the subset of descriptors that corresponds to the current model. This step is repeated until every compound in the training set has been eliminated and its activity is predicted once.
Cross-validated r2 (q2) value can be calculated by using following equation, where yi and y* are the actual and predicted activities of the ith compound, respectively, and ymean is the average activity of all compounds in the training set. Both summations are over all compounds in the training set. The obtained q2value is indicative of the predictive power of the current k-NN QSAR model in predicting compounds in training set.
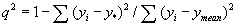
Above procedure is repeated for k=2, 3, 4, etc. Formally, the upper limit of k is the total number of compounds in the data set. The k value that leads to the highest q2 value is chosen for the current k-NN QSAR model.
External validation (pred_r2) using weighted k-nearest neighbor [26]: Following procedure was applied for external validation.
(1) Predict biological activity of a compound in the test set on the basis of the k-NN principle, i.e., as the weighted average activity of k (that correspond k value for highest q2 value) most similar molecules in the training set. The similarities are evaluated as Euclidean distances between compounds using only the subset of descriptors that corresponds to the current model (for highest q2value).
(1) Repeat step 1 for every compound in the test set.
(2) Calculate the predicted r2 (pred_r2) value using following equation, where yiand y* are the actual and predicted activities of the ith compound in test set, respectively, and ymean is the average activity of all compounds in the training set. Both summations are over all compounds in the test set. The obtained pred_r2 value is indicative of the predictive power of the current k-NN QSAR model for external test set.
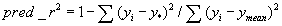
Different training and test set of isatin (1-H-indole-2,3-dione) derivatives were constructed using sphere exclusion data selection method at different dissimilarity levels (3.0 to 5.5). Training and test set were selected using sphere exclusion method at different dissimilarity level if they follow the Uni column statistics, i.e., maximum of the test is less than maximum of training set and minimum of the test set is greater than of training set, which is prerequisite for further QSAR analysis (Table 2). This result shows that the test is interpolative i.e., derived from the min-max range of training set. The mean and standard deviation of the training and test set provides insight to the relative difference of mean and point density distribution of the two sets. k-Nearest neighbor molecular field analysis (kNN-MFA) was applied using stepwise (SW), simulated annealing (SA) and genetic algorithm (GA) approace QSAR models. Results of models developed by SW-kNN MFA, SA-kNN MFA and GA-kNN MFA are shown in Table 3. Best three significant models generated on the basis of predicted correlation coefficient are shown in Table 4. Following statistical measure was used to correlate biological activity and molecular descriptors: n=number of molecules, Vn=number of descriptors, k=number of nearest neighbor, df=degree of freedom, r2=squared correlation coefficient, q2=cross validated correlation coefficient (by the leave-one out method), pred_r2=predictive correlation coefficient for external test set, pred_ r2se=coefficient of correlation of predicted data set, Z score=the Z score calculated by q2 in the randomization test, and α=the statistical significance parameter obtained by the randomization test. Data fitness plot for models 1-3 are shown in Figures 2-4. Result of the observed and predicted biological activity for the training and test compounds in the models 1-3 are shown in Table-5. The plot of observed vs. predicted activity of training and test sets for models 1 to 3 is shown in Figures 5-7. From the plot it can be seen that kNN-MFA model is able to predict the activity of training set quite well (all points are close to regression line) as well as external. Sphere exclusion (SE) algorithm allows constructing training sets covering all descriptor space areas occupied by representative points. Between 3.0 to 5.5 dissimilarity levels which comprises of test set size 4 to 10, kNN-MFA methodology with stepwise (SW), simulated annealing (SA) and genetic algorithm (GA) was used for building the QSAR models. Four predictive models were generated with SW-kNN MFA (pred_r2=0.7552 to 0.9376), three predictive models were generated with SA-kNN MFA (pred_r2=0.7019 to 0.9367) and two predictive models were generated with GA-kNN MFA (pred_r2=0.8226 to 0.8497). The kNN-MFA contour plots (Figures 8) provided further understanding of the relationship between structural features of substituted isatin derivatives and their activities which should be applicable to design newer potential CE inhibitors. Further increase in dissimilarity value has produced decrease in model quality.
| Column Name | Average | Max | Min | StdDev | Sum |
|---|---|---|---|---|---|
| Training set (pKi) | 5.5896 | 8.0969 | 4.0726 | 1.1302 | 245.9431 |
| Test set(pKi) | 5.3106 | 6.6990 | 4.5346 | 0.9228 | 26.5531 |
Table 2: Uni-Column Statistics for model 1 for training and test set activity.
| SW-KNN MFA | SA-KNN MFA | GA-KNN MFA | |||||
|---|---|---|---|---|---|---|---|
| Dissimilarity Value | Test set mol | q2 | pred_r2 | q2 | pred_r2 | q2 | pred_r2 |
| 3.0 | 23;34;35;5 | 0.8272 | 0.8528 | 0.7679 | 0.6081 | 0.7131 | 0.8226 |
| 4.0 | 23;34;35;4;5 | 0.8211 | 0.9376 | 0.7348 | 0.7200 | 0.7673 | 0.8497 |
| 4.5 | 23;33;35;40;43;5;55 | 0.8221 | 0.8906 | 0.7522 | 0.7019 | 0.6585 | 0.6785 |
| 5.0 | 17;23;33;34;43;6;40;55; 47 | 0.8346 | 0.7552 | 0.8038 | 0.5230 | 0.6228 | 0.7101 |
| 5.5 | 17;23;33;34;43;5;6;40;55;47 | 0.9012 | -0.1341 | 0.6662 | 0.0375 | 0.7961 | 0.3852 |
Table 3: Result of kNN-MFA study using various variable selection methods.
| Parameters | Model-1 (Dissval=4.00) SW-kNN MFA | Best Model-2 (Dissval=3.00) SA-kNN MFA |
|---|---|---|
| Training Set Size (n) | 44 | 42 |
| Test set size | 5 | 7 |
| k nearest neighbour | 3 | 3 |
| q2 | 0.8211 | 0.8221 |
| q2se | 0.4780 | 0.4830 |
| pred_r2 | 0.9376 | 0.8906 |
| pred_r2se | 0.2433 | 0.3048 |
Table 4: Statistical significant models generated using kNN-MFA method on the basis of predictive correlation coefficient (Best three model).
| S. No. | Compound | Actual pKi | Predicted pKi | |
|---|---|---|---|---|
| Model 1 | Model 2 | |||
| 1 | 2 | 4.4179 | 4.2856 | 4.42142 |
| 2 | 3 | 4.4622 | 4.4775 | 4.57359 |
| 3 | 4 | 4.5346 | 4.9248* | 4.80192 |
| 4 | 5 | 5.8013 | 6.1564* | 5.22974* |
| 5 | 6 | 6.1308 | 6.1973 | 5.03719 |
| 6 | 10 | 4.1662 | 4.5053 | 4.68625 |
| 7 | 13 | 6.0223 | 5.366 | 5.96055 |
| 8 | 14 | 6.0605 | 6.2589 | 5.9456 |
| 9 | 15 | 7.4949 | 6.8866 | 7.42994 |
| 10 | 16 | 7.1739 | 7.0584 | 7.49075 |
| 11 | 17 | 5.9666 | 6.3036 | 5.74261 |
| 12 | 18 | 5.5406 | 4.6004 | 6.64539 |
| 13 | 19 | 6.3872 | 6.0876 | 5.76312 |
| 14 | 20 | 6.2147 | 6.6778 | 6.42331 |
| 15 | 21 | 5.5702 | 6.1764 | 5.61473 |
| 16 | 22 | 6.9586 | 6.4886 | 6.7688 |
| 17 | 23 | 6.699 | 6.8574* | 6.83228* |
| 18 | 24 | 7.3279 | 6.5834 | 6.58432 |
| 19 | 25 | 6.7696 | 6.7559 | 6.77363 |
| 20 | 27 | 5.2588 | 4.5878 | 6.0899 |
| 21 | 28 | 4.4214 | 5.4597 | 4.84025 |
| 22 | 29 | 5.1096 | 4.6401 | 5.14193 |
| 23 | 33 | 4.8268 | 4.5935 | 4.61013* |
| 24 | 34 | 4.8761 | 4.7465* | 5.21946 |
| 25 | 35 | 4.6421 | 4.6747* | 4.61006* |
| 26 | 36 | 5.1255 | 4.399 | 5.13725 |
| 27 | 38 | 6.5528 | 6.8635 | 5.783 |
| 28 | 40 | 4.5272 | 4.9229 | 4.6101* |
| 29 | 43 | 5.0200 | 4.6755 | 5.02772* |
| 30 | 45 | 4.4763 | 5.0249 | 4.49826 |
| 31 | 47 | 4.5114 | 4.9379 | 4.91948 |
| 32 | 48 | 4.0726 | 5.0253 | 4.53604 |
| 33 | 49 | 5.5918 | 6.1975 | 5.92876 |
| 34 | 50 | 6.2076 | 5.8884 | 5.72282 |
| 35 | 51 | 6.1871 | 5.9035 | 5.73006 |
| 36 | 52 | 4.7799 | 5.0287 | 5.0603 |
| 37 | 53 | 5.3893 | 4.7203 | 5.21589 |
| 38 | 54 | 4.6615 | 5.0894 | 5.45793 |
| 39 | 55 | 4.7645 | 4.8172 | 4.36713* |
| 40 | 56 | 4.6615 | 4.6952 | 4.51981 |
| 41 | 57 | 4.2716 | 4.8949 | 4.65049 |
| 42 | 58 | 5.1302 | 4.3986 | 4.94322 |
| 43 | 59 | 4.6536 | 4.6057 | 4.33347 |
| 44 | 60 | 4.5622 | 5.3129 | 4.28558 |
| 45 | 61 | 4.6478 | 5.3305 | 4.51014 |
| 46 | 62 | 8.0969 | 7.9786 | 7.55117 |
| 47 | 63 | 7.9586 | 8.051 | 7.76452 |
| 48 | 64 | 5.8125 | 4.8107 | 5.19885 |
| 49 | 65 | 8.0000 | 7.1532 | 6.89096 |
*Indicates that the compounds are in the test set.
Table 5: Actual and predicted biological activity for models 1-3.

Figure 3: Data fitness plot for Model 1.

Figure 4: Data fitness plot for Model 2.

Figure 5: Graph between actual and predicted activity for training and test set (Model 1).

Figure 6: Graph between actual and predicted activity for training and test set (Model 2).

Figure 7: kNN-MFA result plot: 3D-alignment of molecules with the important steric and hydrophobic points contributing with ranges of values shown in parenthesis for model 1.

Figure 8: kNN-MFA result plot: 3D-alignment of molecules with the important steric and hydrophobic points contributing with ranges of values shown in parenthesis for model 2.
Interpretation of model 1
It is produced by using stepwise kNN-MFA method having internal predictivity 82.11% (q2=0.8211) and external predictivity 93.76% (pred_r2=0.9376). kNN-MFA plot shown in Figure 8. This model showed that hydrophobic (H_977) and steric (S_619) interactions play important role in determining carboxyl esterase inhibitory activity. Hydrophobic field descriptor (H_977) has positive range (0.3552 to 0.7596) indicates that positive hydrophobic potential is favorable for increase in activity and hence more hydrophobic substituent group is preferred in that region. Compounds having lesser hydrophobic substituent group are not favorable for biological activity. Steric field descriptor (S_619) has negative range (-0.0599 to -0.0386) indicates that negative steric potential is favorable for increase in activity and hence less bulky substituent group is preferred in that region. Compounds having more bulky substituent group is not favorable for biological activity.
Interpretation of model 2
It is produced by using stepwise kNN-MFA method having internal predictivity 82.11% (q2=0.8211) and external predictiviy 89.0% (pred_ r2=0.89). kNN-MFA plot shown in Figure 4. This model showed that hydrophobic (H_986) and steric (S_620) interactions play important role in determining carboxyl esterase inhibitory activity. Hydrophobic field descriptor (H_986) has positive range (0.2413 to 0.2947) indicates that positive hydrophobic potential is favorable for increase in activity and hence more hydrophobic substituent group is preferred in that region. Compounds having lesser hydrophobic substituent group are not favorable for biological activity. Steric field descriptor (S_620) has negative range (-0.0624 to -0.0277) indicates that negative steric potential is favorable for increase in activity and hence less bulky substituent group is preferred in that region. Compounds having more bulky substituent group is not favorable for biological activity.
In conclusion, the model developed to predict the structural features of Isatins (Indole-2,3-diones) to inhibit carboxylesterases reveals useful information about the structural features requirement for the molecule. In optimized models, Model 1 is giving very significant results. The master grid obtained for the various kNN-MFA models show that negative value in electrostatic field descriptors indicates the negative electronic potential is required to increase activity and more electronegative substituents group is preferred in that position, positive range indicates that the group which imparts positive electrostatic potential is favorable for activity so less electronegative group is preferred in that region. Negative range in steric descriptors indicates that negative steric potential is favorable for activity and less bulky substituents group is preferred in that region. Positive value of steric descriptors reveals that positive steric potential is favorable for increase in activity and more bulky group is preferred in that region. On the basis of the spatial arrangement of the various shapes, electrostatic and steric potential contributions model proposed in this work is useful in describing QSAR of Isatins, Indole-2,3-diones derivatives as carboxylesterase inhibitors and can be employed to design new derivatives with more potent inhibitory activity.