Journal of Physical Chemistry & Biophysics
Open Access
ISSN: 2161-0398
ISSN: 2161-0398
Research Article - (2015) Volume 5, Issue 6
The geometric, thermodynamic, electronic and absorption properties of Pyrrole and some of its derivatives have been carried out using CCSD/6-311++G(d,p)/STO-3G, TD-DFT and DFT/B3LYP/6-31G(d) from monomer to five repeating units. Substitution by a methyl group at C3 and functional groups at C4 cause small changes in atomic distances. The estimated inter-ring bond length based on Badger's rule of 1.41 Å indicates that the average structure is about 30% quinoid. The geometries indicates that strong conjugate effects and effective aromatic structure are formed in the order Pyrrole>MPCam>MPC. The oligomers of simulated compounds have been extrapolated to polymer through second-degree polynomial-fit equation with r2 value ranging from 0.96-0.99. Calculated band gap of pyrrole, which is 2.9 eV, significantly correlates with the experimental value which ranges from 2.9-3.2 eV and this corresponds to π-π* transition energies. Natural bond orbitals of polypyrrole reveals that the wavefunctions contain dynamic correlations (single reference), closed shell character while substituted polypyrrole are multireference (static correlation), open shell character.
<Keywords: Polypyrrole; Natural orbitals; Multireference; Band gap
The applications of conducting polymers have been extensively studied over the years due totheir electronic, electrochemical and optical properties. Polymers that could effectively interact with biological systems and applied in molecular electronics, sensors and surgical plasters [1-6] have attracted much interest.
Conducting polymers have been classified based on their method of synthesis, conduction and nature. Based on conduction, electroactive polymers have been classified based on redox polymers and electronconducting polymers [7-8]. Polypyrrole has been classified as electronically conducting Polymer that contains different redox active groups [9]. In electronically conducting polymers, the motion of delocalized electrons occurs through conjugated systems however, the electron hopping mechanism is likely to be operative, especially between chains (interchain conduction) and defects.
Polypyrrole has been synthesised by various methods of chemical and electrochemical polymerizations and it is among the most widely studied conducting organic polymers, experimentally and theoretically, this is due to their chemical stability, high conductivity upon doping [10]. Novel approach has been implemented in order to study biological electron transport between polymeric materials and proteins using functionalized conducting polymers to produce thin films which has been used for the covalent binding of electrode surfaces to glucose oxidase [11].
Gooding et al. have fabricated the glassy carbon electrode modified with anti-rabbit IgG antibody entrapped in an electrodeposited polypyrrole membrane for label free amperometric detection of rabbit IgG antigen in flow injection system [12]. Farace et al. have developed a reagentless immunosensor for the detection of luteinising hormone based on antibody entrapped in a conducting polypyrrole matrix using impedance spectroscopy [13]. Also in recent years, various enzyme immobilization matrices such as polyrrole were used in construction of amperometric cholesterol biosensors. polypyrrole doped with polyvinyl sulfonate [14] to detect urea were recently used.
Barker et al. and Kudera et al. [15,16] reported the existence of reversible electron transfer between electrodes and peptide proteins, this has opened interesting viewpoint in terms of developing conducting polymers for biosensor devices. Polymers of 3-methyl pyrrole-4-carboxylic acid (MPC) has been synthesized and applied as a biosensor for cytochrome c. However no voltametric response was observed with the polymer of unsubstituted pyrrole and this was ascribed to the exclusion of the electron withdrawing carboxylic acid group which appears to be necessary for electron transfer.
This research work is geared towards understanding the molecular, stability and reactivity properties of polymers of pyrrole and its substituted analogues. The thermodynamic properties, especially enthalpy of formation and zero point energy are vital to establish structural, energetic and reactivity relationships. These properties are useful in chemistry, medicine, and industries [17]. These properties are necessary in evaluating the stability, reactivity and nature of reactions.
Nero et al. [18] carried out a theoretical examination on the geometries and spectroscopic properties of MPC oligomers and some of its related parent’s structures. The geometry was determined using semi-empirical (AM1, PM3) methods, while spectroscopic properties were investigated using ZINDO-S/CI.
In this work Density Functional Theory (DFT) has been used to estimate the geometries, enthalpy of formation, dipole moments and polarizabilities of studied systems. Some spectroscopic parameters have been investigated using Time Dependent Density Functional Theory (TDFT). Coupled Cluster Theory (CCSD) has been used to ascertain if the ground state is dominated by non dynamic (static) correlation (arising from near-degeneracies among electronic configuration) or dynamic correlations (arising from coulomb repulsion). T1 diagnostics and Natural bond orbital coupled cluster tool calculation have also been carried out. These methods are useful and reasonably accurate tools for the exploration of the spectroscopic and geometric properties of conjugated polymers.
This paper is ordered as follows: Section 2 explains the theory used, section 3 provides details of the computational methods used, section 4 shows the results and discussion, and conclusions are presented in section 5.
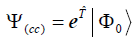 (1)
(1)
Where Ψ(cc) is the CCSD wave function, where Φ0 is the uncorrelated Hartree−Fock wave function and the operator 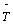 = T1 +T2 represents the CCSD amplitudes.
= T1 +T2 represents the CCSD amplitudes.
Time dependent Density Functional Theory (TDDFT)
TDDFT is a quantum mechanical approach in physics and chemistry to survey the properties and dynamics of many-body systems in the presence of time-dependent potentials, such as electric or magnetic fields. TDDFT is a conservatory of DFT but provides exact and basically suitable methods to evaluate electronic excitation energies of isolated systems and less commonly, solids [19].
The formal foundation of TDDFT is Rung-Gross (RG) theorem [20], which is the time dependent analogue of Hohenberg-Kohn (HK) theorem [21].
The Rung and Gross approach considers a single component systems in the presence of time –dependent scalar field where the Hamiltonian is represented in the form.
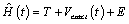 (2)
(2)
Where T is the kinetic energy operator, E is the electron-electron interaction, Vexact(t) the external potential which defines the system in as well as number of electrons. The many-body wavefunction start off from the time-dependent Schrödinger equation.
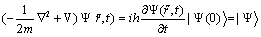 (3)
(3)
In elucidation, the rung-Gross theorems utilizing the Schrödinger equation is the ab initio
The Kohn-Sham formalism has been very much thriving in ground state DFT. Its time dependent stability looks alike in the logic that it chooses a non-interacting in which to form the density that is equal to the interacting system. This is based on the fact that the non-interacting systems can be derived and the wavefunction can be represented as a Slater determinant of single-particle orbitals.
The difficulty is to determine a potential, denoted as Vs(r,t) or VKS(r,t), which in turn give rise to a non-interacting Hamiltonian, Hs,
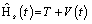 (4)
(4)
This determines a wavefuntion
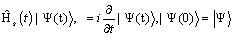 (5)
(5)
Which is constructed in terms of a set of N orbitals which obeys the equation,
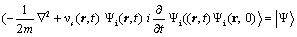 (6)
(6)
and generate a time-dependent density ρs
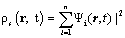 (7)
(7)
Where ρs is the density of the interacting system.
The test is determining the approximations to Kohn-sham potential. The time-dependent KS potential is toppled to exact the external potential of the system and the time-dependent Coulomb interaction, VJ. The component left is the exchange-correlation potential
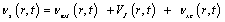 (8)
(8)
Computational details
The geometries of polypyrrole and substituted polypyrrole have been optimized with RB3LYP/6-31G(d) level of theory as shown in Figure 1. The optimized geometries of studied systems are included in the supplementary data. Calculations on the ground state geometry, energy orbitals, energy gaps, dipole moments, thermodynamic properties and energies have been investigated with DFT methods. Excitation energies, oscillatory strength and UV/VIS absorption wavelength have been determined using TDDFT. The heat of formation has been determined using Semi-empirical methods. T1 diagnostics and Natural Bond Orbital (NBO) was carried using Coupled Cluster Single and Double (CCSD) methods at 6-311++G(d,p) and STO-3G level of theoryAll calculations have been carried out on the optimized ground state structures using Spartan 10 and Guassian 09 software packages on a 2.50 GHz, dual core i5-2450M CPU personal computer.

Figure 1: Simulated Enthalpies as a function of the number of monomer unit.
Molecular geometry
The geometric properties studied at ground state were selected bond length, bond angle, dihedral angle (torsional angle) and the intermolecular charge transfer (ICT). These were determined using restricted hybrid Density Functional Theory (DFT) method at B3LYP/6-31G*.
Bond Length Alternation parameter (BLA): In order to characterize the alternation of single or double carbon-carbon bonds within the two ring moieties, the Bond Length Alternation parameter (BLA) has been used [22]. This parameter might be defined as the average of absolute values of the differences between the i-th bond length di and average bond length 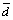 and N stands for the number of conjugated C—C bonds.
and N stands for the number of conjugated C—C bonds.
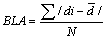 (9)
(9)
Inter-ring bond length: The inter-ring bond length has been also determined using Badger's rule [23]. The estimated inter-ring bond length based on Badger's rule is 1.42Å indicating that the average structure is about 30% quinoid like. Cuff and M. Kertesz, determined bond distances through the application of Badger's rule.
Accordingly,
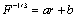 (10)
(10)
Where F is the stretching force constant in mdyn/A°, r is the C-C bond length in Å, and a and b are constants
Intramolecular charge transfer (ICT): This has been calculated as the average of the summation of Mulliken charge distribution of each studied system.
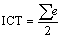 (11)
(11)
Where e represent summation of mulliken charge on each atom
Thermodynamic properties
Thermodynamic properties considered in this study include: Calculated energies (E0) Zero Point Energy (ZPE), Enthalpy of formation (ΔH°f), Entropy (ΔS) and Heat of formation (ΔH°f). These have been determined using restricted hybrid Density Functional Theory (DFT) method at B3LYP/6-31G*. The Heat of formation (ΔH°f), was calculated using Semi-empirical methods (MNDO, AM1, PM3 and PM6)
Percentage difference in stability
The percentage difference in the stability of titled compounds were determined using the following expression
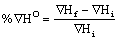 (12)
(12)
ΔH°f = Enthalpy change after adding the substituent
ΔH°i = Enthalpy change before adding the substituent
Electronic properties
The Ionization potential (IP), Electron affinity (EA), HOMO, LUMO, and band gap calculations have been determined on these optimized structures. The oligomers were extrapolated to polymer through second-degree polynomial fit equation [24]. The band gap (or the π−π* lowest electron transition) is estimated as the difference between the HOMO and LUMO orbital energies. The negative of HOMO energy is estimated as IP, [25,26] whereas the negative of LUMO is estimated as EA [27]. All calculations have been performed in the gas phase.
Determination of hardness/ softness
Based on Koopman’s theorem [28], the ionization energy, electron affinity and global electrophilicity index ω: introduced by [29] has been determined using the following expression
Where;
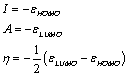 (14)
(14)
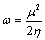 (15)
(15)
Electronic transition properties
The detailed electronic transitions, including excitation energies, oscillator strength, MO/character (configurations for the main S0→S1 electronic transitions) and maximum absorption in UV-Vis spectrum (denoted as λmax) are presented in this work. For UV−VIS calculations, TDDFT calculations have been performed at B3LYP/6-31G*
Geometric properties
Geometric properties of PyrroleL: The C−C bond length are found to be in the range of 1.378 to 1.425 Å, the C−H bond length was 1.082 Å, the C−N bond length is 1.375 Å and the N−H bond is 1.01 Å. The C−C−C is 107.7° (107.4°), H-N-C bonds are 125.1° (125.1°) and those of C−N−C are 107.7 to 109.8° (107.4°). The dihedral angles of C−C−N-C are in the range of 0.03 to 0.79°. The calculated bond angles of pyrrole are all similar to the experimental value as seen in Table 1
| Pyrrole | MPC | MPCAM | Expt | |
|---|---|---|---|---|
| Selected bonds (Å) | ||||
| N1-C2 | 1.375 | 1.384 | 1.381 | |
| C2-C3 | 1.378 | 1.375 | 1.378 | 1.370a |
| C3-C4 | 1.425 | 1.442 | 1.439 | 1.432a |
| C4-C5 | 1.378 | 1.387 | 1.387 | |
| C5-N1 | 1.375 | 1.358 | 1.364 | 1.349a |
| Selected angles | ||||
| N1-C3-C5 | 107.7 | 107.7 | 107.8 | 107.7b |
| C2-C3-C4 | 107.7 | 108.6 | 106.4 | 107.4b |
| H2-N1-C3 | 125.1 | 125.1 | 125.1 | 125.1b |
| Selected dihedral angles | ||||
| N1-C2-C3-C4 | 0 | 0 | -0.37 | |
| C3-C4-C5-N1 | 0 | 0 | -0.33 | |
| δD(Å) | 0.0154 | 0.021 | 0.0198 | |
| LB (Å) | - | 1.457 | 1.478 | |
| DICT (e) | 0.001 | 0.002 | 0.004 | |
Expta30, b31dD=BLA; LB=inter-ring bond length; DICT=intramolecular charge transfer
Table 1: Selected bond length, bond angle and dihedral of studied compounds.
The C-C bond lengths (atoms C2-C3; 1.378 Å) are closer to experimental value (1.370) [30], Atoms C3-C4 is about 0.007Å shorter than experimental value (I.432), atoms C5-N1 is about 0.026Å longer than experimental value (1.349) as shown in Table 1.
For Pyrrole ring, the summation in equation (9) above runs over the bonds N1—C2, C2—C3, C3—C4, C4—C5, C5—N1 with N = 5. With respect to this definition, a very small BLA value indicates an effective aromatic structure. As it can be seen in Table 1, the BLA value is 0.0154(Å), which is in agreement with that predicted by [22] (0.009- 0.032(Å)).
Intramolecular charge transfer (ICT) was calculated as the average of the summation of Mulliken charge distribution of studied compounds. For pyrrole, the Intramolecular charge transfer value is 0.001e.
The tortional angle calculated for pyrrole is 0° Eclipsed bonds are characterized by a torsion angle of 0° which shows that pyrrole is a planar molecule due to delocalization of the pi electrons and the nitrogen lone pair. The planarity of the chain structure is shown to be important for high conductivity in pyrrole polymers which appears to involve spinless carriers perhaps associated with bipolarons.
Geometric properties of MPC: The C−C bond length is found to be in the range of 1.375 to 1.442 Å, the C−H bond length is 1.082 Å to 1.098Å, C−N bond length is 1.370 Å to 1.384 Å, and the N−H bond was 1.010 Å to 1.023 Å. The C−C−C is 107.7 to 108.6° and those of C−N−C are 107.7 to 111.36°.The dihedral angles of C−C−N-C is 0°
Substitution of hydrogen atom in pyrrole ring by a methyl group at C3 (in 3-methylpyrrole) induces fairly small changes in ring inter atomic distances. C-C bond lengths of MPC are closer to the experimental value of pyrrole.
The C3-C4 bonds was about 0.01Å longer than the experimental value of pyrrole (I.432) and 0.017Å longer than the calculated value. Atoms C5-N1 was about 0.009Å longer than experimental value (1.349) and 0.017 Å shorter than the calculated value. Substitution of hydrogen atom in pyrrole by a methyl and carboxylic group at C3 and C4 results to a slight increase in C−C−C bond angle as compared to the experimental value of pyrrole (107.4°) as seen in Table 1.
In MPC, C3-C4 (1.442 Å) and C4-C5 (1.387Å) bond lengths are longer, and weaker than the C3-C4 and C4-C5 (1.425 and 1.378 Å respectively) bonds for pyrrole. This is due to the electron-withdrawing character of the carboxyl group and electron releasing methyl group which affect the π-conjugation in the pyrrole ring, leading to some asymmetries and changes in the strength of the adjacent bonds to the atoms in which these groups are attached and, consequently, an alteration of the bond lengths.
The Bond Length Alternation parameter (BLA)was also calculated to be 0.021(Å) and for MPC, which is slightly higher than that of pyrrole and this is in agreement with the value obtained for pyrrole as stated by Rottmannova et al. The mulliken charge distribution for modeled compound was also obtained, which gave a higher DCT than pyrrole compound, indicating a higher redistribution of electron. The dihedral angle obtained for MPC is 0° which also shows a planar structural arrangement as that of pyrrole.
Geometric properties of MPCam
For MPCam, the C−C bond length range from 1.378 to 1.439 Å, the C−H bond length is 1.082Å and the C−N bond length is 1.364 Å to 1.381 Å. The C−C−C is 106.4°, C−N−C is 107.8 to 109.70° and that of H2-N1-C3 is 125.1°.The dihedral angles of C−C−N-C are in the range of -0.33 to -0.37°
The calculated C-C bond lengths of MPCam are closer to the experimental value of pyrrole, but there are no experimental values for this compound. The C3-C4 bond is about 0.007Å longer than the experimental value of pyrrole (I.432). The C4-C5 is about 0.01Å shorter than experimental value (1.397). C3-C4 (1.439 Å) and C4-C5 (1.387Å) bond lengths in MPCam are longer, and hence the bonds are weaker than in pyrrole; This due to the fact that the substitution of hydrogen atom by an amide group at C4 (CONH2) brings about a deactivation effect. When the amine group is attached to the ring through the carbonyl carbon, the partial positive charge on the carbon removes electron from the ring leading to a weakening of the bonds.
The Bond Length Alternation parameter (BLA) was also calculated to be 0.0198Å which is similar to that of pyrrole and also in agreement with the value obtained for pyrrole by Rottmannova et al. The DCT of MPam was found to be 0.003e and 0.0022e higher than pyrrole and MPC respectively, which indicates high intramolecular charge transfer and electron redistribution.
Thermodynamic properties of Pyrrole, MPC and MPCam
Thermodynamic properties of Pyrrole: Zero-point energy (ZPE), also called quantum vacuum zero-point energy, is the lowest possible energy that a quantum mechanical physical system may have; it is the energy of its ground state. All quantum mechanical systems undergo fluctuations even in their ground state and have associated zero-point energy, a consequence of their wave-like nature. The uncertainty principle requires every physical system to have a zeropoint energy greater than the minimum of its classical potential well. ZPE is also a measure of the thermodynamic stability, the more positive the ZPE, the more stable the polymer [32]. Values of ZPE in Table 2, indicates the higher the number of monomers the more stable is the polymer (from 216.89 to 879.43 KJ/mol).
| No of Monomers | ΔHof (kJ/mol) | ZPE (kJ/mol) | ΔSof (J/molo) | ΔGof (kJ/mol) |
|---|---|---|---|---|
| Pyrrole | ||||
| 1 | -210.08 | 216.89 | 270.52 | -210.11 |
| 2 | -418.99 | 383.33 | 346.37 | -419.03 |
| 3 | -627.91 | 548.54 | 418.70 | -627.95 |
| 4 | -836.82 | 714.48 | 480.49 | -836.87 |
| 5 | -1045.73 | 879.43 | 540.34 | -1045.79 |
| MPC | ||||
| 1 | -435.45 | 328.8 | 357.92 | -435.49 |
| 2 | -874.67 | 608.7 | 490.31 | -874.733 |
| 3 | -1311.42 | 887.8 | 610.7 | -1311.49 |
| 4 | -1748.19 | 1168.1 | 723.13 | -1748.27 |
| 5 | -2184.96 | 1451.2 | 826.57 | -2185.06 |
| MPCam | ||||
| 1 | -418.04 | 362.86 | 362.24 | -418.08 |
| 2 | -834.93 | 676.36 | 493.76 | -834.98 |
| 3 | -1251.81 | 987.09 | 620.16 | -1251.88 |
| 4 | -1668.7 | 1301.49 | 730.91 | -1668.79 |
| 5 | -2085.58 | 1613.87 | 845.11 | -2085.68 |
Table 2: Thermodynamics properties of studied systems.
Analyzing the results of the thermodynamic function listed in Table 2 we see that the ΔH° and ΔG° obtained using DFT and B3LYP levels are close. The negative value obtained indicates that the reaction is exothermic. Since ΔG° is negative it is also an indication that the process is spontaneous.
Entropy (S) is a thermodynamic property which originates from the second law of thermodynamics. This property is particularly useful in determining the spontaneous direction of a process and for establishing maximum possible efficiencies. From the result obtained in Table 3, the standard entropy (ΔS°) is positive, and the entropy increases as the number of monomer also increases. The calculated entropy using DFT functional is in agreement with experimental value obtained for pyrrole as shown in Table 2.
| No. of monomers |
EA | IP | ELUMO | EHOMO | Band gap |
|---|---|---|---|---|---|
| (eV) | (eV) | (eV) | (eV) | (eV) | |
| Pyrrole | |||||
| 1 | -1.39 | 5.48 | 1.39 | -5.48 | 6.87 |
| 2 | -0.39 | 4.82 | 0.39 | -4.82 | 5.21 |
| 3 | -0.02 | 4.57 | 0.02 | -4.57 | 4.57 |
| 4 | 0.16 | 4.48 | -0.16 | -4.48 | 4.32 |
| 5 | 0.31 | 4.40 | -0.31 | -4.40 | 4.09 |
| ∞ | -1.20 | -4.12 | 2.92 | ||
| R2 | 0.99943 | 0.99935 | |||
| MPC | |||||
| 1 | 0.23 | 5.93 | -0.23 | -5.93 | 5.7 |
| 2 | 1.1 | 5.91 | -1.1 | -5.91 | 4.81 |
| 3 | 1.56 | 5.61 | -1.56 | -5.61 | 4.04 |
| 4 | 1.53 | 5.51 | -1.53 | -5.51 | 3.98 |
| 5 | 1.58 | 5.11 | -1.58 | -5.11 | 3.53 |
| ∞ | 1.89 | 4.42 | -1.89 | -4.42 | 2.53 |
| R2 | 0.96643 | 0.865146 | |||
| MPam | |||||
| 1 | 0.01 | 5.83 | -0.01 | -5.83 | 5.82 |
| 2 | 0.82 | 5.21 | -0.82 | -5.21 | 4.39 |
| 3 | 0.92 | 5.11 | -0.92 | -5.11 | 4.19 |
| 4 | 0.95 | 4.68 | -0.95 | -4.68 | 3.73 |
| 5 | 1.07 | 4.21 | -1.07 | -4.21 | 3.14 |
| ∞ | -1.07 | -3.45 | 2.38 | ||
| R2 | 098398 | 0.83826 | |||
| aExpt. | 2.9-3.2 | ||||
| a[35] | |||||
Table 3: IPs, EAs, HOMOs, LUMOs and Band Gaps of Studied systems.
Enthalpy (ΔH°) is the thermodynamic function that accounts for heat flow in a system. Thermodynamic stability is expected when ΔH is negative, and the more negative the value of ΔH, the more stable the compound [32]. The value obtained range from -210.8 to -1045.73KJ/ mol. There is an increase in ΔH° as the number of ring increases.
Thermodynamic properties of MPC
The calculated ZPE values for MPC range from 328.8 kJ/mol to 1451.2 kJ/mol as the number of ring increases as shown in Table 2. This value is higher than the calculated ZPE of pyrrole (216.9 kJ/mol), therefore indicating a higher thermodynamic stability.
Enthalpy change (ΔH°f) and free energy (ΔG°f) have closely related values as shown in Table 2. The ΔG°f of MPC was found to be -435.5kJ/ mol which also increases negatively from as the number of ring increases. This is lower than that of pyrrole which is 210.11kJ/mol; this decrease is due to the substitution of hydrogen atom with carboxylic group at the C-4 position and methyl group at the C-3 position. A spontaneous formation between the protein and conducting polymer is expected in MPC than in pyrrole since ΔG°f is more negatively. The values of ΔS° (Table 2.) also increase positively as the number of ring is increased as also seen in pyrrole.
The ΔH°f per ring increases as the number of rings increases (as seen in Table 1; supplementary data) indicating that there is an effective electronic stabilization as expected from the pi electron character of these structures.
The enthalpy change for MPC range from -435.5 to -2184.9kJ/mol as the number of ring is increased (Table 2). Thermodynamic stability is expected when ΔH°f is negative, and the more negative the value of ΔH, the more stable the polymer compound [33]. MPC is more stable thermodynamically than pyrrole since it has a more negative ΔH°f value. Statistically, it was observed there was a significant difference at p< 0.05 level between ΔH°f values of pyrrole and MPC, and also a 107% increase in the stability was also observed.
Thermodynamic properties of MPCam
The result shown in Table 2 reveals a higher ZPE values for MPCam as compared to MPC.
The results shown in Table 2 indicate that the enthalpy change (ΔH°) and free energy (ΔG°) are closely related and have higher values than those of MPC. The ΔG° values have been found to be in the range -418.1 to -2085.7kJ/mol which also decreases as the number of ring is increased. These results obtained show a spontaneous process for the formation of MPCam.
Result shows that ΔH°f of this substituted pyrrole is lower than the ΔH°f of pyrrole, and higher than that of MPC. The negative ΔH°f value of MPCam obtained indicates an exothermic reaction and as well as a spontaneous process as compared to pyrrole. This change attributed to the proximity of the amide group attached to the ring, which removes electronic density from the ring. The ΔH° obtained for MPCam range from -418.0 to -2085.6kJ/mol as the number of ring is increased as shown in Figure 1. A significant difference was also observed between pyrrole and MPCam at p
Molecular energies of Pyrrole: The IP and EA of an atom or a molecule are the energy needed to remove or gain an electron and therefore can be referred to as the ability of the molecule to donate and accept electrons. Table 3 shows the IP and EA of pyrrole. Table 3 shows that all of the IP values from monomers up to five repeating units are positive, whereas some of the EA values are in negative. The negative EA indicates that the anionic state is unbound. It can be seen that most HOMO energies vary in the range of -4.4 to approximately -5.48, while LUMO energies vary significantly (from -0.31 to 1.39). As shown in Figure 2, we find a linear relationship between the direct calculated vertical HOMO energies and 1/n calculated from DFT (with a correlation coefficient r2 = 0.99) and the direct calculated vertical LUMO energies and 1/n calculated from DFT (with a correlation coefficient r2 = 0.99). The LUMO; of π-nature is delocalized over the C-C bonds of the pyrrole ring, while the HOMO is located over the C=C bonds (Figure 3)

Figure 2: Simulated plot of HOMO, LUMO energy level against 1/n of oligomer.

Figure 3: The frontier molecular orbital of pyrrole.
Energy gap between the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO), or simply HOMO-LUMO gap (HLG; ΔEg), also called Band gaps, is a key parameter because is a measure of the electron density hardness [34].
The band gap of 5 pyrrole oligomers is extrapolated to polymer through second-degree polynomial fit equation. The experimental band-gap value of pyrrole ranging 2.9-3.2 eV, according to Zotti et al. [35] significantly correlates well with the calculated band gap of pyrrole, which is 2.9 eV, and this corresponds to π-π* transition energies (Table 3).
A series of studies have shown that a small band gap corresponds to a high reactivity and with anti-aromaticity. According to [36], absolute hardness (half of HOMO/LUMO energies) is commonly used as a criterion of chemical reactivity and stability. They pointed out that aromatic rings influence the reactivity through aromatic π-electrons delocalization of positive charge and also increasing aromaticity results in an increase in hardness and the decrease in reactivity. A more reactive molecule is characterized by a lower value of μ, η. Calculation shows that polypyrrole exhibits high reactivity, low stability, antiaromatic behavior and it is term a soft molecule. (Supplementary data)
Molecular energies of MPC
The results for IP and EA of MPC are presented in Table 3. Result also shows that all values of IPs and EA from monomers up to five repeating units are positive values. The HOMO energies vary within the range of -4.42-5.93, while LUMO energies vary significantly (from -0.23 to -1.89). Figure 2, shows a linear relationship between the direct calculated vertical HOMO energies and 1/n calculated from DFT (with a correlation coefficient r2 = 0.87) and the direct calculated vertical LUMO energies and 1/n calculated from DFT (with a correlation coefficient r2 = 0.96). The LUMO extended over the C-C bonds, the nitrogen and oxygen atom, this involve delocalization over the entire molecule framework while the HOMO is located over the C=C bonds (Figure 4)

Figure 4: The frontier molecular orbital diagram HOMO (-5.2 eV) of MPC.
The band gap of MPC oligomers was also extrapolated to polymer through second-degree polynomial fit equation. A significant decrease in band gap of 2.9eV to 2.53eV was observed for MPC. This difference is due the substitution of hydrogen atom in by a methyl group at C3 and the electron-withdrawing character of the carboxyl group which affects the π-conjugation in the pyrrole ring. Increase conjugation also contributes to the decrease in energy gap. This agrees with experimental results and it’s comparable to the work of Guimaraes et al. [37]. According to [28-30], MPC exhibits high reactivity, low stability, antiaromatic behavior and termed as a soft molecule as characterized by a lower value of band gap, μ and η as shown in supplementary data, when compared with pyrrole. This also implies that the interaction and binding effect of polymer to the protein will be more spontaneous compared to that in pyrrole.
Molecular energies of MPCam
Calculated results for IP and EA of MPCam are shown in Table 3. This reveals that all values of IPs and EA from monomers up to five repeating units are also positive values. The HOMO energies vary within the range of -3.45 to -5.83, while LUMO energies vary significantly (from -0.01 to -1.07). As shown in Figure 2, there is a linear relationship between the direct calculated vertical HOMO energies and 1/n calculated from DFT (with a correlation coefficient r2 = 0.84) and the calculated vertical LUMO energies and 1/n calculated from DFT (with a correlation coefficient r2 = 0.98). The LUMO frontier molecular orbital extended over the C-C bonds, the nitrogen and oxygen atom, this involve delocalization over the entire molecule framework while the HOMO is located over the C=C bonds (Figure 5)

Figure 5: The frontier molecular orbital diagram HOMO (-4.6 eV) of MPCam.
The band gap of 5 MPC oligomers was also extrapolated to polymer through second-degree polynomial fit equation. The band gap is obtained from the difference of the orbital energies (valence and conduction band). A significant decrease in band gap from 2.9eV to 2.38eV was observed for MPCam. The substitution of hydrogen atom by an amide group at C4 (CONH2) brings about a deactivating effect. The amide group been attached to the ring through the carbonyl carbon, brings about a partial positive charge on the carbon and has as a deactivating influence which also affects the π-conjugation in the pyrrole ring. Increase conjugation also contributes to the decrease in energy gap. This result is also comparable to the work of Guimaraes et al. MPCam will exhibits a higher reactivity, lower stability, antiaromatic behavior and termed as a softer molecule as characterized by a lower value of band gap, μ and η as shown in supplementary data, when compared with pyrrole, and MPC.
Electronic Absorption Properties
The extent of absorption of a new material determines how it can orient the protein and enable electron transfer. This is an important factor in biosensor application. A good biosensor material should have broad and strong visible absorption characteristics.
On the basis of the optimized ground-state structures, we present, the excitation energy Eex(eV), calculated absorption λmax (nm) and oscillator strength (O.S) along with main excitation configuration of the oligomers of studied systems. These values are calculated using TDDFT method starting with optimized geometry B3LYP/6-31G(d) level.
Electronic Absorption Properties of Pyrrole
The simulated absorption spectrum of pyrrole is presented in Figure 6. The calculated wavelength λmax (nm) increases as the number of ring increases. The largest oscillator strengths (O.S) originate from S0 → S1 electronic transition. Excitation to S1 state corresponds exclusively to the promotion of an electron from the HOMO to the LUMO. As in the case of the oscillator strength, the absorption wavelength arising from S0 → S1 electronic transition increases progressively with the increase in the conjugation. As seen in Table 4, the bands signed at 190.59, 239.29, 279.44, 306.27, and 329.52 (nm) corresponds to the HOMO-LUMO transition and is of ICT (intramolecular charge transfer) character thus possessing averagely high transition intensity.

Figure 6: Calculated UV-Vis spectra of studied systems.
| No. of Monomers | Electronic transition | λmax (nm) | E(exc)(ev) | O S | MO/Character | Coefficient |
|---|---|---|---|---|---|---|
| 1 | S0→S1 | 190.59 | 6.51 | 0.00 | HOMO→LUMO | 0.999 |
| S0→S2 | 180.87 | 6.86 | 0.0044 | HOMO→LUMO+1 | 0.65 | |
| S0→S3 | 172.98 | 7.17 | 0.1940 | HOMO→LUMO-1 | 0.94 | |
| 2 | S0→S1 | 239.29 | 5.18 | 0.6932 | HOMO→LUMO | 0.97 |
| S0→S2 | 221.37 | 5.60 | 0.0001 | HOMO→LUMO+1 | 0.89 | |
| S0→S3 | 211.52 | 5.86 | 0.0009 | HOMO-1→LUMO | 0.64 | |
| 3 | S0→S1 | 279.44 | 4.44 | 0.9818 | HOMO→LUMO | 0.99 |
| S0→S2 | 253.12 | 4.90 | 0.0092 | HOMO→LUMO+1 | 0.81 | |
| S0→S3 | 238.99 | 5.19 | 0.0014 | HOMO→LUMO+2 | 0.93 | |
| 4 | S0→S1 | 306.27 | 4.05 | 1.0534 | HOMO→LUMO | 0.99 |
| S0→S2 | 277.90 | 4.46 | 0.0142 | HOMO→LUMO+1 | 0.78 | |
| S0→S3 | 251.79 | 4.92 | 0.0238 | HOMO→LUMO+3 | 0.89 | |
| 5 | S0→S1 | 352.76 | 3.76 | 0.9379 | HOMO→LUMO | 0.99 |
| S0→S2 | 308.91 | 4.17 | 0.0373 | HOMO→LUMO+1 | 0.76 | |
| S0→S3 | 284.61 | 4.62 | 0.4973 | HOMO→LUMO | 0.67 |
Table 4: Electronic transition data obtained by the TD/DFT-B3LYP/6-31G(d) calculation for pyrrole.
Absorption Properties of MPC
The simulated absorption spectrum of MPC is presented in Figure 6. The calculated wavelength λmax (nm) also increases as the number of ring increases. The largest oscillator strengths (O.S) also originate from S0 → S1 electronic transition. Excitation to S1 state involves the promotion of an electron from the HOMO to the LUMO. As in the case of the oscillator strength, the absorption wavelength arising from S0 → S1 electronic transition increases progressively with the increase in the conjugation. As seen in Table 5, the bands signed at 235.05, 276.55, 317.70, 345.36, and 376.51 (nm) corresponds to the HOMO-LUMO transition and is of ICT (intramolecular charge transfer) character thus possessing higher transition intensity. Also it is has been established that organic molecules (including biological molecules and conjugated molecules) have a strong triplet-triplet transition [38,39]. For two, three, four and five rings MPC the singlet-singlet transition basically involves four (|H>, |H-1>, |L>and |L+1>) but preliminary calculations of spectra show an increase of the transition levels which can be used to orient the molecule and enable electron transfer.
| No. of Monomers | Electronic transition | λmax (nm) | E(exc)(ev) | O.S | MO/Character | Coefficient |
|---|---|---|---|---|---|---|
| 1 | S0→S1 | 235.05 | 5.28 | 0.0455 | HOMO→LUMO | 0.31 |
| S0→S2 | 233.76 | 5.30 | 0.0003 | HOMO-1→LUMO | 0.99 | |
| S0→S3 | 200.24 | 6.19 | 0.2639 | HOMO→LUMO | 0.89 | |
| 2 | S0→S1 | 276.55 | 4.48 | 0.4035 | HOMO→LUMO | 0.97 |
| S0→S2 | 268.21 | 4.62 | 0.0405 | HOMO→LUMO+1 | 0.96 | |
| S0→S3 | 251.66 | 4.93 | 0.0047 | HOMO-1→LUMO | 0.65 | |
| 3 | S0→S1 | 317.70 | 3.90 | 0.4236 | HOMO→LUMO | 0.981 |
| S0→S2 | 290.57 | 4.27 | 0.2339 | HOMO-1→LUMO | 0.23 | |
| S0→S3 | 276.21 | 4.49 | 0.0286 | HOMO→LUMO+2 | 0.97 | |
| 4 | S0→S1 | 345.36 | 3.59 | 0.4789 | HOMO→LUMO | 0.98 |
| S0→S2 | 320.70 | 3.87 | 0.3947 | HOMO→LUMO+1 | 0.95 | |
| S0→S3 | 303.01 | 4.09 | 0.1178 | HOMO→LUMO+2 | 0.95 | |
| 5 | S0→S1 | 380.51 | 3.29 | 0.7449 | HOMO→LUMO | 0.97 |
| S0→S2 | 353.42 | 3.51 | 0.0698 | HOMO→LUMO+1 | 0.96 | |
| S0→S3 | 345.94 | 3.58 | 0.0627 | HOMO→LUMO+2 | 0.96 |
Table 5: Electronic transition data obtained by the TD/DFT-B3LYP/6-31G (d) calculation for MPC.
Absorption Properties of MPCam: The absorption spectrum of studied compounds as seen in Figure 6 reveals that the largest oscillator strength and absorption wavelength also originates from S0 → S1 electronic transition, these increases progressively with the increase in the conjugation. As shown in Table 6, the HOMO-LUMO transition bands are 251.55, 300.40, 312.63, 365.67, and 372.47nm. In comparison with the maximum absorption of (MPC) (376.51nm), there is a difference about 4.04 nm for (MPCam) (372.47nm), which is attributed to the low ICT. However, transition distributions have shown that in all cases for MPCam the |H>orbital contains significant contributions to the spectra.
| No. ofMonomers | Electronic transition | λmax (nm) | Eexc (ev) | O.S | MO/Character | Coefficient |
|---|---|---|---|---|---|---|
| 1 | S0→S1 | 251.55 | 4.93 | 0.0012 | HOMO→LUMO | 0.77 |
| S0→S2 | 232.49 | 5.33 | 0.0376 | HOMO+1→LUMO | 0.92 | |
| S0→S3 | 203.50 | 6.10 | 0.2019 | HOMO-1→LUMO | 0.75 | |
| 2 | S0→S1 | 300.40 | 4.13 | 0.4614 | HOMO→LUMO | 0.97 |
| S0→S2 | 274.03 | 4.53 | 0.0177 | HOMO→LUMO+1 | 0.94 | |
| S0→S3 | 263.01 | 4.71 | 0.0041 | HOMO-4→LUMO | 0.75 | |
| 3 | S0→S1 | 312.63 | 3.97 | 0.5506 | HOMO→LUMO | 0.97 |
| S0→S2 | 294.74 | 4.21 | 0.1948 | HOMO→LUMO+1 | 0.98 | |
| S0→S3 | 269.29 | 4.60 | 0.0595 | HOMO-1→LUMO | 0.67 | |
| 4 | S0→S1 | 365.67 | 3.39 | 0.6315 | HOMO→LUMO | 0.99 |
| S0→S2 | 334.87 | 3.70 | 0.2365 | HOMO-1→LUMO | 0.91 | |
| S0→S3 | 323.22 | 3.84 | 0.1734 | HOMO→LUMO+1 | 0.91 | |
| 5 | S0→S1 | 380.63 | 3.33 | 0.9062 | HOMO→LUMO | 0.98 |
| S0→S2 | 347.85 | 3.60 | 0.0334 | HOMO→LUMO+1 | 0.78 | |
| S0→S3 | 344.46 | 3.66 | 0.1027 | HOMO→LUMO+3 | 0.75 |
Table 6: Electronic transition data obtained by the TD/DFT-B3LYP/6-31G (d) calculation for MPCam.
Open or close shell nature of studied systems
Inorder to ascertain if the ground state is dominated by non dynamic (static) correlation (arising from near-degeneracies among electronic configuration) or dynamic correlations (arising from coulomb repulsion), T1 diagnostics and Natural bond orbital [40] coupled cluster tool was carried using Coupled Cluster Single and Double (CCSD) methods at 6-311++G(d,p) and STO-3G level of theory. T1 diagnostics (< 0.02) indicates that the system is single reference (closed shell), while (>0.02) indicates that the system is multireference in nature (open shell).
An easy way to ascertain whether there are unpaired electrons in a correlated wave function is to examine the occupation numbers of the spinless natural orbitals; in a closed-shell configuration, these are always 2 doubly occupied or 0 unoccupied, while values close to 1 indicate single occupancy and unpaired electrons [41]. In Figure 7 we plot the occupancies of the natural orbitals for the studied systems. We have designated the two orbitals with occupancies closest to 1 the highest occupied natural orbital HONO with occupancy greater than 1 and lowest unoccupied natural orbital LUNO with occupancy less than 1, respectively.

Figure 7: Natural orbital occupation numbers for the near singly occupied orbitals of studied systems as a function of chain length using CCSD/STO-3G basis.
These natural orbitals together with usual highest occupied molecular orbital HOMO and lowest unoccupied molecular orbital LUMO are shown in Figure 7 The result shows that poly pyrrole is a single reference(dynamic correlation), closed shell system while other studied systems are multireference (static correlation), open shell in nature (see supplementary data).
The geometric, thermodynamic, electronic properties and Absorption properties have been carried out carried out using Quantum mechanical calculation. Pyrrole and its derivatives have been studied using CCSD/6-311++G(d,p), STO-3G level of theory, TD-DFT and DFT B3LYP/6-31G (d) level of theory from monomer up to five repeating units.
The mutual comparison of the obtained equilibrium geometries showed that the DFT predicted C-C and C=C bond length are in agreement with the experimental values. In order to characterize the alternation of single or double carbon-carbon bonds within the two ring moieties, the Bond Length Alternation parameter (BLA) was used. The BLA values were also in agreement with the experimental values. However, substitution of hydrogen atom in pyrrole by a methyl group at C3 (in 3-methylpyrrole) and other functional groups at C4 causes fairly small changes in ring inter atomic distances. The estimated inter-ring bond length based on Badger's rule is 1.41Å indicating that the average structure is about 30% quinoid. The calculated geometries indicate that the strong conjugate effects and effective aromatic structure are formed with the order as Pyrrole>MPCam> MPC. Substitution of some functional groups into the backbone of pyrrole structure led to an increase effective electronic stabilization. The enthalpy change of MPCam indicates that it is more thermodynamically stable and has a more effective electronic stabilization, while unsubstituted pyrrole has the lowest. The IP, (EA), HOMO, LUMO, and band gap of optimized structures have been calculated. The oligomers of simulated compounds were extrapolated to polymer through second-degree polynomial fit equation with r2 value ranging from 0.66-0.99. Calculated band gap of pyrrole, which is 2.9 eV, significantly correlates with the experimental band-gap value which ranges from 2.9-3.2 eV and this corresponds to π-π* transition energies.
The substitution of methyl and some functional groups destabilized the HOMO and LUMO levels, decreased the band gaps, and increased the conjugation length, which are essential features in the design of low band gap conducting polymers. The substitution of COOH on the backbone of pyrrole (MPC) lowers the LUMO energy; this is due to electron localization. The order of energy gap is MPCam< MPC <pyrrole In general calculated values of Eg, μ, η and ω, according to koopman’s theorem and Fukui function f(r), which are reactivity descriptors lead to the conclusion that MPCam is more reactive, lower stability and aromatic behaviour and termed as a softer molecule than other studied compounds. The UV-Vis absorption spectra have been simulated by TD-DFT calculations. The oscillator strength and the absorption wavelength arising from S0 → S1 electronic transition; increases progressively with the increase in the conjugation. Excitation to S1 state also corresponds to the promotion of an electron from the HOMO to the LUMO. In comparison with the maximum absorption of studied compounds, MPCam has the highest (395.05nm), which is attributed to the strong intramolecular charge transfer. Analysis of the Natural bond orbitals reveals that pyrrole is a single reference (dynamic correlation), closed shell system while substituted polypyrrole are multireference (static correlation), open shell in nature.
It can be concluded that in the polymeric form of systems, the molecular orbitals reveal that certain transitions are allowed and certain transitions are forbidden this is due to the symmetry of the ground and exited state. These properties suggest these compounds (MPCam< MPC) to be good polymers for biosensor applications. We hope that our results may provide a reference for further experimental and theoretical work as well as effects directed towards the synthesis and application of these derivatives.
The authors are grateful to University of Ibadan and CV Raman fellowship for the grant. CU would like to acknowledge Dr Debashree Ghosh for making her lab available for use in India.