Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2018) Volume 11, Issue 12
Background: Finding accurate genome structural variations (SVs) is important for understanding phenotype diversity and complex diseases. Limited research using classification to find SVs from next-generation sequencing is available. Additionally, the existing algorithms are mainly dependent on an analysis of the alignment signatures of paired-end reads for the prediction of different types of variations. Here, the candidate SV regions and their features are computed using single reads only. Classification is used to predict the variation types of these regions.
Results: Our approach utilizes reads with multi-part alignments to define a possible set of SV regions. To annotate these regions, we extract novel features based on the reads at the breakpoints. We then build three random forest classifiers to identify regions with deletions, inversions, or tandem duplications.
Conclusions: This paper proposes a random forest-based classification approach, MPRClassify, which addresses the issue of finding SVs using single reads only. These single-reads are used to define candidate regions and extract their features. Experimental results show that single reads are sufficient to find SVs without the need for paired-end read signatures. Our proposed approach outperforms existing approaches and serves as a basis for future studies finding SVs using single reads.
Keywords: Structural variation; Next generation sequencing; Split read; Single read; Random forests; Supervised learning
A genomic structural variation (SV) is defined as a rearrangement of a genome region at least 50 base pairs (bps) long [1]. There are several forms of SVs, including insertion, deletion, inversion, copy number variation (CNV), and translocation. SVs have a huge impact on the phenotype and genotype of complex diseases such as Mendelian and cancers [2-4]. Thus, much effort has been put into finding these variations from next-generation sequencing (NGS) datasets. However, finding genome structural variations in whole genome sequencing (WGS) is a difficult task, and solutions are still in the early stages. Various methods have been developed to find structural variations from WGS data generated by NGS platforms such as Illumina and Ion Torrent. These datasets are known as paired-end reads; a pairedend read refers to two short sequences that are generated from DNA fragment ends. A typical approach to SV identification begins with aligning paired-end reads to a reference sequence. The read alignment signals are then analyzed to predict SVs. Alkan et al. divided these signals into four classes: read-pair (RP), split-read (SR), read-depth (RD), and assembly-based (AS) [1]. RP-based algorithms analyze abnormal paired-end read alignments that are not mapped as expected in terms of distance, orientation, or order of read-ends [5-8]. SR-based methods are used to find the best partial alignment for unmapped reads in detecting sequences of SV breakpoints at single-nucleotide precision [9]. RD-based approaches analyze read count changes between genome regions to find copy number variations [10-12]. AS-based approaches either use assembly graphs to call SVs [13] or refine SV breakpoints as a post-processing stage for other approaches [14]. In fact, no single signal can find all SV types and sizes with accurate breakpoints. Thus, recent studies combine two or more signals [15]. Because of the great contribution of SR-based approaches in finding accurate SV breakpoints, several approaches utilize SR with other signals. In Delly [16] and Prism [17], RP signals are used to predict the types and regions of SVs. Subsequently, unmapped read-ends are divided into splits and realigned to the putative breakpoint regions. Delly and Prism differ in their search spaces for breakpoints. Additionally, read aligners have improved to support partial alignment for reads that are difficult to align completely. In the literature, these reads are known as softclipped reads (SCR). BWA-MEM [18] and Bowtie2 [19] are examples of such aligners. Accordingly, many SV calling algorithms use RP and SCR analysis. Usually, RP signals are used to predict candidate SV regions and their types, followed by an SCR analysis for refining. For example, CREST [20] assembles the SCRs at a breakpoint and maps the longer sequence into the reference genome to find other breakpoints. In Softsv [21], SCRs inside potential SV regions are aligned to each other to find precise breakpoints. Conversely, Softsearch [22] uses SCRs for breakpoint prediction of potential SVs first, then searches for the RP signals around the breakpoints for predicting variation type. Manta [23] incorporates RP signals, SCR signals, and AS into two scoring models for germline and somatic variations. Svelter [24] uses RP signals and SCR to generate potential breakpoints that are clustered according to their positions. Then, the breakpoints of two adjacent clusters are rearranged and scored based on the insert size, orientation, and read depth for each rearranged structure. Limited research has been conducted investigating the use of supervised learning techniques for finding SVs. Both SVM2 [25] and ForestSV [26] compute their features based on RP and RD signals. While SVM2 uses a multi-class SVM with polynomial kernel for classifying insertion and deletion regions, random forests are used in ForestSV to find duplications and deletions. Neither SVM2 nor ForestSV were designed to predict accurate breakpoints. Alternatively, Wham [27] uses RP, SR, and SCR signals to predict initial SVs regions, which are then classified by a random forest classifier. Svclassify [28] uses one-class support vector machines to classify SV regions and non-SV regions using features derived from multiple sequencing technologies. Classification-based approaches use different techniques to extract and annotate candidate SV regions. Moreover, the training samples for them are different due to the lake of a real benchmark data for SVs identification problem. Existing methods mainly use information extracted from RP signals in order to predict SVs. RP information includes: distance between readends, strands of read-ends, and order of read-ends. In this study, we investigate the use of single-reads information only without requiring RP information for predicting SVs. This is achieved by developing a multi-part read alignment classifier (MPRClassify) to find deletions, inversions, and tandem duplications. MPRClassify uses clipped reads to define potential SV regions. The regions are annotated with 61 features based on clipped reads, RD, and rearrangement of breakpoint regions. We trained three independent random forests (RF) [29] using simulated training sets. The classification models are applied to simulated and real datasets and MPRClassify performance is compared to five state-of-the-art approaches.
Given a set of read alignments as a BAM file format [30] and a reference genome sequence to which the reads are aligned, MPRClassify computes the genome’s mean coverage and extracts the clipped reads, defines the set of potential SV regions (PSV), extracts features for these regions, and builds three random forest models to identify the regions of deletions, inversions, and duplications. The main workflow of MPRClassify is shown in Figure 1. MPRClassify assumes that read alignments are generated by a read aligner that supports multi-part alignment for reads that cannot be aligned completely to the reference sequence.

Figure 1: MPRClassify workflow. The potential SVs regions are extracted. The random forests (RF) models are built by using simulated SVs regions (orange path). The models are used for predicting SVs from new samples (blue path).
Extracting clipped reads
In this stage, the BAM file containing the alignments is parsed to extract clipped reads (CRs). A CR is a read-end that has been aligned to a reference sequence with a clipped portion from at least one of its ends because of sequencing errors or crossing SV breakpoints. Figure 2A-C shows the clipped reads at breakpoints of deletion, inversion, and tandem duplication. According to the SAM/BAM specification [31], clipped reads are recorded either as soft-clipping or hard-clipping. In soft-clipping, the complete read sequence is kept, while only the mapped portion is kept in the case of hard-clipping. Most read aligners mark the top hit alignment of the read parts as soft-clipped and the alignments of other parts as hard-clipped. In our approach, we use the BWA-MEM [18] aligner to align the reads to the reference sequence. It supports the multi-part read alignment, and we recommend using it with MPRClassify. MPRClassify assumes that each clipped-read creates a potential SV's breakpoint at the position where the read has been clipped (br1 and br2 in Figure 2). A clipped-read CR is defined as a 9-tuple including: read name/ID, reference name where the read was aligned, breakpoint position on the reference sequence, alignment strand, length of the clipped portion, mapping quality of alignment, coverage of left region of the breakpoint (L in Figure 2D), coverage of right region of the breakpoint (R in Figure 2D), and read sequence. The length of the left (L) and right (R) regions is given as a threshold (covRegLen); we use 100 bps, as shown in Figure 2D. Region coverage is the average number of times the region has been mapped by reads. The read alignments used for computing coverage should be more than minimum mapping quality (minMapq); MPRClassify uses 30 as the default value. MPRClassify ignores the clipped reads that have a clipped portion smaller than the minimum clipped sequence length (clipLen); 10 bps is used as a default value for clipLen. Moreover, MPRClassify only considers read alignments that are not flagged as duplicate PCR and pass quality control. The final outputs of this stage are two sets of soft-clipped reads only (SCR) and hard-clipped reads only (HCR).

Figure 2: An illustration for the single reads that are aligned at breakpoints of deletions (a), inversions (b), and tandem duplications (c). Red is used for the reads that are aligned to the forward strand and the blue color for reads aligned to the other strand. LRs and RRs are the reads clipped at the start and end regions of SV. In (d) we illustrate the regions (L and R) that are used for computing coverages around breakpoints.
Generation of potential SV regions
In this step, potential SV regions (PSV) are defined by pairing SCR breakpoints and HCR breakpoints. The clipped-pair scri ∈ SCR and hcrj ∈ HCR forms a potential SV region psvk ∈ PSV if and only if scri and hcrj are parts of the same read and are aligned to the same reference. To find deletions, inversions, and duplications, clipped-pairs with different references are ignored. The clipped-read with the lower breakpoint position represents the left-read (LR) that creates br1 and the right- read (RR) that creates br2. Therefore, each psvk ∈ PSV is defined as a 15-tuple of: reference name/ID, position of the LR (br1), position of the RR (br2), strand of LR, strand of RR, coverage of L region at br1, coverage of R region at br1, coverage of L region at br2, coverage of R region at br2, the soft clipped-read location (at br1 or br2), the side of the clipped part of LR (left or right), the side of the clipped part of RR (left or right), deletion-score, duplication-score, and inversionscore. The last three scores are the pairwise sequence alignment scores of the sequence alignment of scri to the rearranged breakpoints regions generated by respectively applying deletion, duplication, and inversion. The pairwise sequence alignment score is the largest score of the best matches between two sequences. Alignment score calculation depends on the alignment type (local, global, or hybrid), substitution scoring schema, and gap penalties. MPRClassify uses the local alignment algorithm (Smith-Waterman) [32] to align reads to the rearranged regions. The scores are then divided by the length of scri sequence. MPRClassify ignores the PSV regions, which are less than 50 bps or overlap gap regions. The final PSV are passed to the next stage for computing classification features.
Feature extraction
Given a PSV set from the previous step, MPRClassify defines 61 new binary features (0 or 1) for each psvk ∈ PSV as follows.
• Features 1–28 are for coverages around breakpoints (L and R regions). The coverage values are converted from numbers into nominals. Each coverage value is represented by seven nominal values.
• Features 29–44 are for the differences of coverages at breakpoints (between L and R regions at both br1 and br2). Each value is converted into eight nominal values.
• Features 45–47 are for the number of other entries in PSV sharing both br1 and br2, sharing at least br1, and sharing at least br2. These values should exceed a defined threshold (supportThr). MPRClassify uses half of the genome's mean coverage as a default for this threshold.
• Features 48–51 are for the strands of the read parts that form the PSV entries. The first feature represents whether the read parts are aligned in the same strand. The other features are for the other entries in PSV with parts mapped on different strands and sharing both br1 and br2, sharing at least br1, and sharing at least br2. As in features 45–47, these values should exceed supportThr.
• Features 52–57 are extracted from the alignment scores. The first three features represent whether the alignment scores exceed a defined threshold (alignThr). The last three features encode the largest score.
• Features 58–61 are for encoding the order of read’s parts that form a PSV entry.
• Feature 62 is the class of structural variation of the region. For deletion classifier, the value of this attribute is either deletion or not-deletion. The same way is used for labeling examples for inversion and duplication classifiers.
To remove highly correlated features, the Phi-coefficient is used to measure associations between attributes. The Phi-coefficient was introduced by Karl Pearson to measure correlations between binary variables [33]. If the absolute correlation between two attributes is above a threshold, the attribute with the largest mean absolute correlation is removed. All filtered features are recorded in an ordered vector and used as a feature set for building and testing random forest classifiers.
Random forests
Random forest was introduced by Breiman for classification and regression [29]. It is an ensemble of classification trees [34] in which each tree is built using a bootstrap sample of the training samples and a random selection of features at each split. For classification, predictions are made by a majority vote of the trees while averaging their outputs in regression. In our context, a random forest model is trained to predict structural variation type (feature 62). By using the feature set from the previous stage, binary random forest classifiers are trained using simulated training examples. Random forest models are trained for binary classification for deletions, inversions, and duplications. To find the optimal classifiers, the classifiers are trained using different configurations of the number of trees (1, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000) and the number of variables at each split (4–20). Accordingly, we train 187 models for each SV class (deletions, inversions, and duplications).
Training dataset
Simulated datasets are used to train three classifiers for the three SV types: deletion, inversion, and tandem duplication. To generate training examples, a set of 1000 deletions, 1000 inversions, 500 insertions, and 1000 tandem duplications was introduced in a copy of the human reference genome hg19 using RSVsim [35]. RSVsim is a simulator that simulates SVs of different types and sizes. Subsequently, paired-end reads are generated from the rearranged genome by wgsim [36]. We set the wgsim parameters (insert size, standard deviation read length, and base error rate) to 250, 75, 100, and 0.001. The reads are then aligned in the reference genome by BWA-MEM. The median coverage of the genome is 5x. Finally, the PSV regions and their features are computed as described in the Methods section. The default values are used for the thresholds. To train the models, the PSV regions are labeled first. The class of a psvk is set to deletion only if psvk has a reciprocal overlapping with a deleted region in the ground truth with a breakpoint divergence between 0−50 bps from the actual breakpoints. Inversion and tandem duplication regions are labeled similarly. The PSV contains 10,060 regions (942 deletions, 6112 duplications, 1963 inversions, and 1043 others). We use 0.75 for filtering attributes, as the threshold and feature set are reduced to 40 features.
Testing dataset
We simulated 15 whole genomes with the same variations using different combinations of read lengths and read depths. As in the training sample, we use RSVsim to generate variations in chromosomes 1–22 of the human reference genome hg19. The number of variations is 1500 for each SV class (deletion, inversion, and tandem duplication). wgsim and BWA-MEM are used to generate testing BAM samples. The insert size of the reads is 500 for all samples. The read lengths are 75, 100, and 150, and the read coverages are 5x, 10x, 15x, 20x, and 25x. In addition to the simulated dataset, we applied MPRClassify to the whole genome of the real sample NA12878. We chose this sample because it has high-quality benchmark SV calls [28]. The ground truth contains 2,593 deletions in chromosomes 1–22. We only consider deletions of at least 50 bps. The minimum and maximum SV length in the ground truth are 50 bps and 139,620 bps, respectively. Small deletions (50–500 bps) are the dominant group in the ground truth (about 70%). The NA12878 alignments are obtained from the 1000 Genomes Project [37], a low-coverage sample (about 5x coverage mean) with an average read-length of 101 bps. The alignments were generated by BWA-MEM aligner and in CRAM format. Subsequently, it was converted to BAM format to be processed by MPRClassify and other SV calling tools. SAMtools [30] is used for format conversion. The reference sequence for the sample is GRCh38. Because of the difference between the reference genome of the alignments (GRCh38) and the ground truth (hg19), the coordinates of the ground truth were converted to GRCh38 using the UCSC batch coordinate conversion (liftover) [38].
Performance evaluation
We evaluate MPRClassify’s performance using the simulated and real datasets. We use sensitivity, precision, specificity, and F-score as defined in equations 1–3.
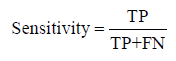 (1)
(1)
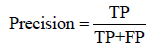 (2)
(2)
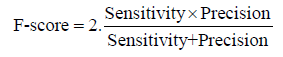 (3)
(3)
Where TP is the number of correctly identified SVs, FP is the number of non-SVs that were incorrectly identified, and FN is the number of SVs that were incorrectly rejected. In this work, a prediction is assumed as a positive if it overlaps an SV in the ground truth of the same type and the divergence breakpoint distance is up to 50 bps. MPRClassify is benchmarked in both simulated and real datasets. The sensitivity, precision, and F1-score are used to compare MPRClassify with five state-of-the-art SV identification tools, namely Delly (version 0.7.6) [16], Softsv (version 1.4.2) [21], Manta (version 1.4.0) [23], Svelter [24], and Wham [27]. Only Wham use a classifier for predicting SV classes. Although SVM2 [25] and ForestSV [26] are classificationbased approaches, we exclude them from the comparison because they do not utilize clipped-reads/split-reads in determining breakpoints of SVs. Their predictions are not accurate at lower base level. In our comparison, we are interested in evaluating prediction at lower base level resolution. The proposed solution and all approaches that are included in the comparison are either using SCR, SR, or both to find precise location of SVs. The default settings are used for running MPRClassify, Delly, Softsv, Manta, Svelter, and Wham across the simulated dataset. Figure 3 shows the overall MPRClassify performance in terms of F-score using models with different combinations of the number of trees and number of variables at each split. The best performance is achieved at 400 trees and four variables for deletions, one tree and 14 variables for inversions, and 300 trees and four variables for duplications. The performance of individual genomes can be seen in the supplementary file. Table 1 and Figure 4 present the performance comparison of MPRClassify against state-of-the-art approaches on simulated datasets. For finding deletions, MPRClassify’s performance is comparable to Delly, Softsv, and Manta; however, MPRClassify achieves the highest precision (99%). Given the low coverage genomes (<15x) with read length (≥ 100 bps), MPRClassify is the best choice for finding accurate deletions. To find tandem duplication, MPRClassify scores the highest performance (97% F-score and 99% precision). For finding inversions, the results show that Softsv is better than the rest of the algorithms. MPRClassify has comparable performance to Delly and Manta and outperforms Svelter and Wham. To benchmark MPRClassify on the real sample NA12878, the comparison is performed for deletions only due to the lack of accurate SV calls for other SV types. MPRClassify, Delly, Softsv, Manta, and Wham were run in their default modes. To run Svelter, the parameter null-copyneutral-perc was set to 0.5, as recommended by Svelter’s authors, for use with low coverage genomes less than 10x. We assume that an SV prediction is a true positive if it overlaps with a ground truth region and if the breakpoint divergence distances of the actual breakpoints are within 50 bps. Table 2 shows that MPRClassify outperforms the other approaches in terms of overall performance (51% F-score) and precision (82%). Delly has the highest sensitivity (49%). Considering SCR-based approaches, MPRClassify has the highest sensitivity compared to Softsv, Manta, Svelter, and Wham. The results also indicate that Delly and our approach are more sensitive when finding small deletions (50–500 bps) and unique true positives over other approaches (Figures 5 and 6). In order to evaluate the performance of MPRClassify in detecting deletions in repeat regions vs. non-repeat regions, we split the ground truth regions according to whether they include overlapping repeats or not. The repeat regions in the reference genome GRCh38 are obtained from the UCSC’s table browser. These regions were generated by RepeatMasker [39]; a tool for finding repeats in DNA sequences. Accordingly, there are 1392 (about 54%) regions with overlapping repeats and 1201 (about 46%) regions without overlapping repeats. Table 3 shows the performance of the compared algorithms in both these regions.
| Deletion | Inversion | Tandem duplication | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Algorithm | Sensitivity | Precision | F-score | Sensitivity | Precision | F-score | Sensitivity | Precision | F-score |
| MPRClassify | 0.855 | 0.990 | 0.917 | 0.937 | 0.917 | 0.927 | 0.940 | 0.991 | 0.965 |
| Delly | 0.927 | 0.947 | 0.937 | 0.952 | 0.902 | 0.927 | 0.902 | 0.987 | 0.943 |
| Softsv | 0.914 | 0.933 | 0.924 | 0.977 | 0.971 | 0.974 | 0.995 | 0.934 | 0.963 |
| Manta | 0.908 | 0.979 | 0.942 | 0.964 | 0.948 | 0.956 | 0.305 | 0.990 | 0.467 |
| Svelter | 0.792 | 0.835 | 0.813 | 0.700 | 0.968 | 0.813 | 0.101 | 0.030 | 0.047 |
| Wham | 0.521 | 0.729 | 0.608 | 0.779 | 0.569 | 0.657 | 0.846 | 0.440 | 0.578 |
Table 1: Sensitivity, precision, and F1-score across the testing simulated genomes.
| Algorithm | #TP | #FN | #FP | Sensitivity | Precision | F1-score |
|---|---|---|---|---|---|---|
| MPRClassify | 969 | 1624 | 214 | 0.374 | 0.819 | 0.513 |
| Delly | 1258 | 1335 | 5353 | 0.485 | 0.190 | 0.273 |
| Softsv | 886 | 1707 | 911 | 0.342 | 0.493 | 0.404 |
| Manta | 731 | 1862 | 194 | 0.282 | 0.790 | 0.416 |
| Svelter | 875 | 1718 | 3050 | 0.337 | 0.223 | 0.268 |
| Wham | 94 | 2499 | 139 | 0.036 | 0.403 | 0.067 |
Table 2: Comparison of NA12878 deletions (at least 50 bps size).
| Algorithm | Regions containing repeats 1392 |
Regions without repeats 1201 |
|---|---|---|
| MPRClassify | 583 (41.88%) | 386 (32.14%) |
| Delly | 740 (53.16%) | 518 (43.13%) |
| Softsv | 525 (37.72%) | 361 (30.06%) |
| Manta | 449 (32.26%) | 282 (23.48%) |
| Svelter | 515 (37.00%) | 360 (29.98%) |
| Wham | 56 (4.02%) | 38 (3.16%) |
Table 3: True predictions in regions containing repeats and regions without repeats in the sample NA12878.

Figure 3: Performance of MPRClassify on simulated dataset using models with different combinations of number of trees and number of variables at each split. (A) Deletion; (B) Inversion; (C) Tandem duplication.

Figure 4: F1-score of MPRClassify and state-of-the-art approaches across testing genomes for deletions (A), inversions (B), and tandem duplications (D).

Figure 5: The pie chart shows the percentage the ground truth (deletions) for NA12878 sample over five size intervals. The bar chart shows the percentage of TP across the five size intervals for NA12878 sample.

Figure 6: The percentage of unique true positives (TPs) of NA12878’s deletions that identified by each approach.
Most existing approaches for finding SVs are based on RP signals for predicting SVs types. Our study aims to use single reads only to find SVs without using RP signals. MPRClassify uses multi-parts read alignments generated by an NGS read-aligner to form a set of possible SV regions. We introduce a new method for annotating these regions. Instead of using an RP signals, MPRClassify uses supervised learning to build three random forest models to classify candidate regions into deletions, inversions, and duplications. The models are trained by a simulated dataset with different combinations of the number of trees and number of nodes at each split. Overall, the proposed approach shows high performance with simulated and real datasets. The average F1-scores are approximately 92%, 97%, and 93% for deletion, duplication, and inversion, respectively. Despite the high precision of MPRClassify, the sensitivity is affected by how candidate SV regions are generated. For deletions, the results of the simulated data indicate that Delly, Softsv, and Manta have higher sensitivity than our approach due to the use of RP signals as a guide for defining breakpoints. While Manta demonstrates the highest performance on the simulated dataset, the results on the real sample (NA12878) do not show the same performance. MPRClassify has the highest F1-score and precision, while Delly has the highest sensitivity (Table 2). Delly’s high sensitivity can be attributed to its method of defining candidate SV regions. Delly realigns both unmapped reads and clipped-reads to refine breakpoints, which increases the number of candidate SVs. However, it is known that approaches which split and realign reads are more sensitive than SCR-based approaches that use clipped-reads generated by the aligners [15]. Wham has the lowest performance for finding NA12878 deletions (Table 2) as a consequence of the low-coverage sequencing of the NA12878 sample (5x average mean coverage). This is confirmed with the results on simulated data (Figure 4). Both our approach and Wham use random forest in classifying candidate regions. However, the results on simulated and real samples confirm that our approach is more robust than Wham (Tables 1-3 and Figure 4). The results on simulated and real datasets confirm that the proposed approach achieves the best performance for finding duplications and deletions in low-coverage genomes with at least 100 bps read length (Figure 4). Our experiments show that single-reads information can be used to predict variation type without using RP information. Our approach achieves the best performance in detecting deletions from low coverage genomes. MPRClassify has three main stages: forming potential SV regions, extracting features of these regions, and applying classifiers for predicting SV class. Therefore, the sensitivity of MPRClassify depends on the sensitivity of generating candidate SVs as well as the classification sensitivity. Generating candidate SV for the NA12878 dataset results in 59% sensitivity, which is higher than Delly (48%). After applying our classifier, the sensitivity is further reduced to 37%. The availability of a real benchmark data for training, would replace the use of simulated dataset and should improve the accuracy of finding SVs using real samples.
In this paper, we introduce MPRClassify, a new approach to finding accurate structural variations from whole-genome sequencing datasets. MPRClassify is based on multi-part read alignments for forming a possible SV set. It is intended to be used for single reads, so it can handle any type of NGS dataset (single or paired-end reads). We define a new method of annotating the candidate regions using clipped-reads, local depth, and rearrangement of breakpoints. For classifying regions, three random forest classifiers are trained by simulated data examples for identifying deletions, inversions, and tandem duplications. Real data experiments and simulations demonstrate that single reads could be used to find SVs without the need for RP signals. We show experimentally that MPRClassify achieves a better performance than state-of-the-art methods for finding deletions in simulated and real samples of low-coverage sequencing.
The authors gratefully acknowledge use of the service of "SANAM" supercomputer at "King Abdulaziz City for Science and Technology" (KACST), Saudi Arabia. The authors also thank Dr. Ghadah Badr for useful discussions at the initial stages of this work.
This research project is supported by a grant from the "King Abdulaziz City for Science and Technology" (KACST), Saudi Arabia (Grant No.1-17-02-001-0024).
Availability of Data and Materials
▪ The alignments file for NA12878 [37] is available at: ftp://ftp.1000genomes. ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/data/CEU/ NA12878/alignment/NA12878.alt_bwamem_GRCh38DH.20150718. CEU.low_coverage.cram
▪ The ground truth SV calls for NA12878 [28] are available at: ftp:// ftp-trace.ncbi.nlm.nih.gov/giab/ftp/technical/svclassify_Manuscript/ Supplementary_Information/Personalis_1000_Genomes_deduplicated_ deletions.bed
▪ The UCSC batch coordinate conversion (liftover) [38] is available at http://genome.ucsc.edu/cgi-bin/hgLiftOver
▪ All the scripts are available upon the request.
EA and AE conceived of the project. EA designed and implemented the work. AE and HA helped in the design and provided expert input. All authors read and approved the final manuscript.
The authors have no conflict of interest to declare.