Journal of Applied Pharmacy
Open Access
ISSN: 1920-4159
ISSN: 1920-4159
Research Article - (2018) Volume 10, Issue 1
A total of 60 peaks generated by forced degradation of three active pharmaceutical ingredients (APIs) were used for the column selection in the development of a stability indicating HPLC-UV analytical method using LC-UV and LC-MS peak tracking. Two mobile phase additives and two organic modifiers were evaluated while screening a list of carefully chosen chromatography columns. The column screening was utilized and the best column selected based on total number of resolved peaks, resolutions, peak widths, and peak shapes. 0.1% TFA in ACN/water was used for the initial screening and optimization of the gradient profile. Three different concentrations of TFA in ACN/water were also evaluated. The optimum TFA concentration, 0.10% (8.77 mM), was considered as optimum for further gradient optimization based on the resolution of critical pairs. After the selection of column, mobile phase and mobile phase modifier (TFA) selection, optimization of the gradient was achieved by a combination of automated chemometric peak tracking and software-based decisions in AutoChrom MS. The correct peak retention equations (i.e., retention time vs. mobile phase ratio) were generated by using first one-step gradients with a wide range of % B followed by optimization in multi-steps gradients. It was found that extrapolation, using quadratic retention models, can lead to large errors in retention time (tR) predictions, especially for poorly-retained components. We present the challenges in resolving the critical resolution pairs, including those with the same m/z, the overestimation and the prediction errors of the software, and why the peaks model (i.e., accuracy of predicted versus experimental) fail at the extremes of the gradient. By using this approach we were able to generate a suitable stability indicating chromatographic method for an extremely challenging sample comprising of three active pharmaceutical ingredients (APIs) and related degradation products with a wide range of hydrophobicity. The APIs were in-house compounds, their identity are blinded in this paper and are not relevant for purpose of the study. There were good matches between the predicted and experimental retention times of the tracked peaks. The peak model was used for the generation of an assay/potency method using the computational tool only.
<Keywords: Analytical method; HPLC; Optimization; Stability indication; Potency assay; Column selection; LC-MS peak tracking; AutoChrom MS
For effective practical method development a combination of screening and optimization should be considered. Most of the stabilityindicating analytical methods in the pharmaceutical industry are gradient reversed-phase HPLC methods. The first step in developing such methods typically involves screening different options for column selectivity [1], mobile phases, and mobile phase additives. This screening process is a key step to getting the desired selectivity. The screening is then followed by optimizing the resolution by fine-tuning the gradient profile and column temperature. Even though this process has been successfully applied to numerous applications, it is accompanied by numerous challenges in each step of method development, such as: column choice for screening, buffer selection, best candidate column selection, etc. [2-5].
Analyte detection using a combination of diode array and mass spectroscopic detectors combined with automation of data acquisition and processing using ACD/AutoChrom MS software (ACD/Labs) has been reported [6-8]. Both Fusion and AutoChrom software were used previously to support HPLC column and mobile phases screening, followed by optimization of the separation using simulation software (ACD/LC Simulator or Drylab) [3]. It was shown that the method development time can be significantly shortened from the typical 1-3 months to about 1 week using method development software [9] and using Quality-by-Design approaches [9]. It is also important to note that the use of systematic method development provides a better understanding of method capabilities and limitations and robustness ensuring a greater chance of successful downstream method validation and transfer. ACD/ Labs improves the performance of HPLC method development by using databases of analytes of a variety of chemical structures that can be used as a starting point for method development [10].
Optimization of the gradient profile is ubiquitous in modern chromatographic method development [11,12]. This is due to the convenience of modification of the gradient profile, as well as its powerful influence on analyte retention. For reversed-phase liquid chromatography, the retention changes in response to changes in the ratio of aqueous mobile phase to organic modifier is well documented. Equation 1 describes the retention factor (k) which is also referred to as capacity factor. The relationship between the retention factor of a given analyte and the volume fraction φ of organic solvent B in the mobile phase is defined by the linear solvent strength relationship, as per Equation 2 [12].
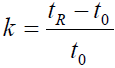 (1)
(1)
Where k is the retention factor, tR is the retention time of the analyte and t0 is the dead time of the column.
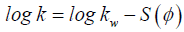 (2)
(2)
where kw is the retention coefficient in water, ϕ is the volume fraction of the mobile phase B solvent, and S is a constant describing the dependence of retention (log k) on solvent ratio (ϕ) for a given analyte and ϕ values is a function of molar mass of the analyte.
The linear solvent strength relationship model does not fit experimental data well at low and and φ values, which may be due to the introduction of different modes of retention such as HILIC [13]. Therefore, polynomials of higher degrees are used to fit the data, as shown in Equation 3. Given the values (C0, C1, and C2), it is possible to model the chromatogram for any isocratic or gradient experiment. Several computer programs have been developed in the mid 90’s either as stand-alone or as part of the system controller or data system [11]. Examples of software packages using this modeling concept are ACD/ LC Simulator, Chrom Sword, and Dry Lab. A similar approach, based primarily on Equation 1, forms the basis of gradient optimization in AutoChrom MS.
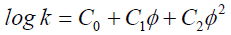 (3)
(3)
where C0, C1, and C2 are constants that depend on the analyte and separation condition.
In gradient elution, the retention coefficient (k*) can be altered by changing parameters such as flow rate, gradient time, change in % organic, and dead volume of the column, as shown in Equations 4 and Equation 5 below. Theoretically, retention time (tR), gradient time (tG), flow rate (F), the change in the volume fraction (Δφ), column dead time (to), peak width at half height (w1/2), k’w (retention time in pure water) can be combined and used to calculate the retention time and peak width at any conditions [14]. The method can be then further optimized by changing the parameters constituting the k* parameter, as per Equation 4. In the contrary to the above approach, method development software uses the retention time of peaks of interest from two gradients of two different slopes at two distinct temperatures to mathematically build the retention time-gradient time - temperature map. The model built by the software is strengthened as more experiments are added and a 3 temperatures x 3 gradients is suggested for a more precise and accurate predictions by the software.
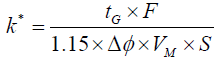 (4)
(4)
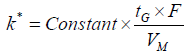 (5)
(5)
where tG is the gradient time, Δϕ is the change in the volume fraction, ϕ is the volume fraction of the mobile phase B solvent during the gradient, F is the flow rate, Vm is the column dead-volume, and S is the slope of the linear relationship log k’ with ϕ.
The present study describes the stability indicating method development for a mixture of 3 APIs and their degradation products, using LC/MS and LC/UV peak tracking with the use of the ACD/ Labs AutoChrom MS software, the challenges in resolving the critical resolution pairs, including those with the same m/z, the overestimation and the prediction errors of the software, and why the peaks model (i.e., accuracy of predicted versus experimental) fail at the extremes of the gradient. We also present how an assay method can be generated using the AutoChrom MS software tool only.
Materials
The three APIs and related authentic by-product and degradation products substances were in-house compounds and their identity is not relevant to this paper. They cover a wide range of hydrophilicities/ hydrophobicities and are representative of common pharmaceutical active ingredients. Acetonitrile (Fisher Scientific, Waltham, MA) and water (Fisher Scientific) were HPLC grade, H2O2 30% (Fisher Scientific), HCl (J. T. Baker, Center Valley, PA), NaOH (Spectrum, Gardena, CA), methanol (EMD, Billerica, MA), trifluoroacetic acid (TFA, J. T. Baker), acetic acid (Sigma-Aldrich, St. Louis, MO) were ACS reagent grade. Buffers at different pHs (pH5 – CH3COOH/CH3COO-, pH 7 and pH 8 – H2PO4-/HPO42-, pH 11 – HPO42-/PO43-) were 10 mM (ionic strength, μ=0.15, adjusted with KCl). The pH 1 was 0.1 M HCl (μ=0.15, KCl).
Acid-base degradation: Forced degradation was performed at pH 1, pH 8 and pH 11 at a concentration of 10 μg/mL drug substance in 10:90 (v/v) acetonitrile/buffer (μ=0.15, KCl). The samples were stored at 80°C for 2 and 7 days for high and low pH, respectively.
Hydrogen peroxide degradation: The oxidation degradation products were generated with 0.1% H2O2 in 90:10 (v/v) phosphate buffer pH 7/acetonitrile, in 60:40:0.2 (v/v) acetonitrile/water/acetic acid and in water. The sample was stored at 80°C for 1 hour or 24 hours, in order to yield sizeable degradation products, and the reaction was quenched by freezing at -20°C.
Composite sample preparation: The composite sample preparation was prepared for two different hydrophobicity regions: (A) low to moderate, in acetonitrile/water 70:30 (v/v) at 100 μg/mL (parent API) and at 0.2 μg/mL (degradation products and process impurities) and (B) high, at 10 μg/mL in 60:40:0.2 (v/v) acetonitrile/water/acetic acid. Both (A) and (B) composites were mixed 1:1 (v/v) to obtain the final composite sample.
HPLC methods: HPLC analysis was performed on an Agilent 1200 instrument controlled by Chromeleon 6.8 software (Dionex, ThermoFisher Scientific, Sunnyvale, CA) for data collection and analysis. Initial HPLC analytical method: The HPLC method used as a starting method for development was able to separate the known impurities on a Zorbax SB-C18 3.5 μ, 3.0 x 150 mm column in a 36 min multi-step gradient. Mobile Phase A: 0.1% (v/v) TFA in water, Mobile Phase B: 0.1% (v/v) TFA in acetonitrile, flow rate 0.8 mL/min, gradient: 10% B (2 min), 10 - 33% B (11 min), 33% B (3 min), 33 - 60% B (13 min), 60 - 80% B (1 min), 80% B (2 min), 10% B (4 min), column temperature 40°C, injection volume 60 μL, UV detection at 270 and 240 nm.
LC/MS Method for column screening: A 5 – 95% B acetonitrile/ water linear gradient with 0.1% TFA was used for initial screening of the suitable column and mobile phase conditions. The column temperature (40°C), injection volume (10 μL) and flow rate (0.5 mL/min) were kept constant. The detection was by UV at 210 and 254 nm, and mass spectrometry (MS). Four columns of different selectivity were screened for the initial column optimization, using the stressed degradation samples: Zorbax C18 column (80 Å, StableBond, SB-C18 USP L1 column, 3.0 x 150 mm, 3.5 μm, Agilent Technologies); Zorbax Phenyl column (80 Å, StableBond SB-Phenyl USP L11 column, 3.0 x 150 mm, 3.5 μm, Agilent Technologies); Atlantis T3 column (C18, 3.0 x 150 mm, 3 μm, Waters Corporation); SunFire C18 column, (3.0 x 150 mm, 3.5 μm, Waters Corporation). The MS data were collected on a Thermo Scientific TSQ Quantum Access triple quadrupole LCMS controlled using the Xcalibur Software. The MS conditions were: +ESI; spray voltage: 4 kV, capillary temperature: 350°C; MS full scan: 100-600 m/z.
LC/MS Method for optimization: The optimization was performed on the Atlantis T3 column for gradient and pH modifier. The mobile phase pH was modified using 0.05, 0.08 and 0.10% (v/v) TFA; however, at higher % TFA, ion suppression made MS tracking of the low intensity ions difficult. Three additional gradients (2 – 4 in Table 1) were run, suggested by AutoChrom software: (1) 5 - 95% B in 60 min, (2) 19 - 95% B in 52 min, and (3) 11-76% B in 42 min. Gradient 1 was chosen as system reproducibility test at the initial and subsequent runs; the latest “optimized” gradient was run and the data added to the model with each set.
| Gradient 1 | Gradient 2 | Gradient 3 | Gradient 4 | ||||
|---|---|---|---|---|---|---|---|
| Time (min) | % B | Time (min) | % B | Time (min) | % B | Time (min) | % B |
| 0.0 | 5 | 0.0 | 5 | 0.0 | 19 | 0.0 | 11 |
| 0.2 | 5 | 0.2 | 5 | 52.0 | 95 | 8.0 | 32 |
| 30.2 | 95 | 60.2 | 95 | 42.0 | 76 | ||
Table 1: Suggested gradients for optimization.
Method development software: The software used for the gradient method development was ACD/AutoChrom MDS LC/MS 12.02, Advanced Chemistry Development, Inc. (ACD), Toronto, Ontario, Canada, further referred to as AutoChrom MS. The AutoChrom MS method development strategy is shown in Figure 1.

Figure 1: Method development strategy.
Automated Data Reduction: One of the largest challenges of rigorous method development is the considerable amount of data to be processed and evaluated. Hyphenated data collection and multivariate method development strategies can result in gigabytes of data for the consideration of the chromatographer. To be practical, method development systems must include the capacity to summarize and extract valuable information from this data as quickly as possible. Figure 2 shows the data reduction process used by the AutoChrom MS software.

Figure 2: Process in AutoChrom MS to extract elution information from LC/MS and LC/UV data files. At various steps in the process, users have the option to review data and make appropriate corrections.
Peak Tracking: Optimization typically uses peak tracking routines to track peaks as parameter changes cause peak retention times to shift. Algorithms have been designed to track components based on spectral shape, mass, peak area, and retention time. In addition, pure standards may be injected where available. In this study, LC/UV and LC/MS data were used in tandem to track peaks from run to run.
UV and MS Mutual Automated Peak Matching (UV-MAP and MS-MAP) are spectral matching techniques designed to match chromatographic peak tables from run to run based on spectral information. While these techniques can be used for both detection and matching, they are normally applied to the matching problem, using peak tables from processing of the Total Absorbance Chromatogram or monochromatic traces to filter the response. The output from the MS-MAP and UV-MAP algorithms for a given component is peak width at half height, asymmetry, and retention time for each peak from each set of conditions.
Detector Reconciliation: The use of multiple detectors (LC/UV and LC/MS) in chromatographic method development provides a higher degree of rigor in detection and tracking of components than individual detectors. While the approach taken in AutoChrom MS relies primarily on LC/MS for peak tracking and LC/UV for chromatographic characteristic of detected peaks, this is not a concrete restriction. Known-mass components can be specifically targeted for tracking (suppressing the LC/UV filtration), and components that fail to ionize can be tracked based on LC/UV spectra.
LC/UV and LC/MS detectors are reconciled in AutoChrom MS using the “critical resolution” concept. In this approach, post-peak tracking results are compared for the detectors, establishing the bestfit matches across the conditions that have been studied. Components that consistently co-elute have a very low critical resolution value, and they are automatically reconciled. Components that have a high critical resolution are retained as unique. This approach rapidly identifies components that have no MS signal. The critical resolution concept has a large advantage over other approaches to reconciling detectors. In cases where there may be a few errors in tracking components, the disagreements are obvious to the system and secondary logic and/or user intervention can be used to fix the problem.
Composite samples: Composite sample is a sample that contains a set of known standards for a given API or set of API’s (n ≥ 2). It has become common practice to perform method development considering components that are not necessarily all part of a single physical sample. Multiple vials can be injected for each set of conditions, and the information combined to form a single overall picture of the elution of all compounds of interest. There are many reasons why this practice is useful. Injecting pure standards or spiked samples can simplify peak tracking. Similarly injecting blanks under each set of conditions can help to identify irrelevant peaks. Also, composite samples can be useful in stability studies, where the subsamples that result from multiple sets of degradation conditions can be individually injected, both easing peak tracking concerns, and avoiding the dilution and other potential concerns that would result from mixing samples prior to injection.
Component reconciliation: The introduction of composite samples to chromatographic method development provides an additional data reduction challenge. This challenge lies in the reconciliation of components from one sample to the next. Any component that is shared between subsamples should be identified as such to avoid errors in decision making. Component reconciliation ideally should be done based on as much information as is possible. AutoChrom MS component reconciliation is done based again on the critical resolution concept. However, in this case spectra are also available from the peak matching results. These spectra are compared. Significant spectral differences will prevent incorrect reconciliation of the components.
Selecting Candidate Systems for Screening: While it may be challenging to interpret the results of a screen when examining column, buffer, and solvent choices, the dramatic effects on sample elution often make these approaches critical to design of excellent chromatographic methods. A great deal of effort has been dedicated to the question of which systems to screen [15-17]. While buffer and solvent choices are relatively limited, there are a huge number of chromatographic columns of different and unique selectivity available to the method development chromatographer. Fortunately many of the most popular reversed-phase chromatographic columns have been characterized based on their retention and selectivity characteristics [18]. A handful of columns can be selected for complementary behavior; these can be incorporated into the method development strategy, reducing the number of experiments while retaining the capacity to readily identify viable systems for optimization. The six chromatographic column parameters of the columns screened and the column descriptors are determined experimentally as described in ref [16]. The retention factor for pentylbenzene, kPB, is largely determined by the surface area and surface coverage (i.e., ligand density). The hydrophobicity or hydrophobic selectivity, αCH2, is the retention factor ratio between pentylbenzene and butylbenzene (αCH2 = kPB/kBB) and is a measure of the surface coverage of the stationary phase as the selectivity between alkyl benzenes differentiated by a single methyl group depends on ligand density. The shape selectivity, αT/O, is the retention factor ratio between triphenylene and the planar related o-terphenyl (αT/O = kT/kO) and is a measure of the shape selectivity, which depends on ligand spacing and the shape/functionality of the silylating reagent. The hydrogen bonding capacity, αC/P, is the retention factor ratio between caffeine and phenol (αC/P = kC/kP) and reflects the number of available silanol groups and the degree of end-capping. The acidic ion-exchange capacity, αB/P at pH 2.7, is the retention factor ratio between benzylamine and phenol at pH 2.7 (αB/P at pH 2.7 = kB/ kP) and is a measure of the acidic activity of the silanol groups. The total ion-exchange capacity, αB/P at pH 7.6, is the retention factor ratio between benzylamine and phenol at pH 7.6 (αB/P at pH 7.6 = kB/kP) and indicates the total silanol activity. The screened column parameters are shown in Table 2.
| Column | kPB | αCH2 | αT/O | αC/P | αB/P at pH 2.7 | αB/P at pH 7.6 |
|---|---|---|---|---|---|---|
| SB-C18 | 6.00 | 1.49 | 1.20 | 0.65 | 0.13 | 1.46 |
| SB-Phenyl | 1.09 | 1.30 | 1.18 | 3.69 | 0.13 | 1.08 |
| Atlantis T3 | 5.48 | 1.47 | 1.17 | 0.49 | 0.11 | 0.25 |
| SunFire C18 | 6.57 | 1.49 | 1.25 | 0.41 | 0.05 | 0.31 |
Table 2: Screened Columns with column selectivity descriptors.
Ranking Screened Systems for Optimizability: The goal of identifying a promising system for optimization typically necessitates a different view of success from optimization. While it is interesting to estimate the suitability of a given method, it is common for all untargeted screening methods to be non-viable for the end use. In addition, some chromatographers will have incomplete information on the sample upon completion of the screening step. It is thus necessary to find a different view of what constitutes “good” and best hit. Many methods have been proposed for quantitative estimation of the quality of screened systems. The simplest of these is a count of the number of resolved peaks. This may be a useful first-pass view, but there are likely better approaches. The resolution score, the count of relevant resolved peaks versus the maximum number observed, is indicative of the average overall resolution between the peaks. The conclusion from a resolution score can be distinctly different from a simple peak count approach. This is due to the fact that AutoChrom MS differentiates between irrelevant and relevant peaks; a peak count approach may include irrelevant peaks in the calculation, meaning that a screening result may appear better than it actually is. It is important to note that some qualities of a system may be practically impossible for software to rank mathematically. For example, a given peak may be of particular interest, and be targeted for higher resolution at the screening stage. Or the width of specific peaks may be considered for detection limit reasons. Any mathematical evaluation of the screened system should be viewed with final method goals in mind – a method with a slightly lower resolution score may be the preferred choice. Visual review of the most promising methods in a screen may very quickly establish an excellent system for optimization that might have been ranked slightly lower by a less flexible algorithm.
All three APIs have vulnerable functional groups that are susceptible to acid, base, peroxide, light and/or thermal stressed conditions. Interaction between the individual degradation products can enhance certain degradation products by Lewis or Brønsted acid or base catalysis. Figure 3 shows the chromatogram of a representative stressed degradation composite sample analyzed by the initial API analytical method. The method separates most of the early eluting degradation peaks; however, there are some overlapping and broad peaks in the 8 – 12 min region (Figure 3a). The top critical resolution pairs shown in Figure 3b, inset were given greater attention for their separation (vide infra). The impurities >24 min in the composite sample were well resolved, but the critical pair at 14.3/14.5 min was not.

Figure 3: HPLC chromatograms in the initial method: (a) Low to moderate eluting peaks at pH 5, 200 hours and 80°C; (b) composite sample with all peaks showing the critical resolution pair (inset right) at 14.3 min/14.5 min.
Column and solvent screen
The initial method used the Zorbax SB-C18 column. In addition to this column we chose three additional columns to evaluate, based on the column selectivity descriptors. In order to build an initial model/ algorithm for each method condition, linear gradients of 5 – 95% B in 30, 40 and 60 min were run on the forced degradation samples containing a total of 60 peaks using the 4 columns (Atlantis T3, Zorbax SB-C18, Zorbax Phenyl, SunFire C-18) and 3 mobile phase systems (ACN/water w/ 0.1%TFA, MeOH/water w/ 0.1% TFA and ACN/water w/ 0.1% HCOOH). Of the three mobile phases, MeOH/0.1% TFA and ACN/0.1% HCOOH were not selected due to low resolutions and peak shapes (data not shown). Although we collected the UV data (in addition to the MS data) for two wavelengths (210 and 254 nm), we chose the 254 nm for further runs. The AutoChrom MS could track 60 peaks on the Zorbax Phenyl column and 57 peaks on the Atlantis T3 column. One could argue that the Zorbax SB-C18 column leads to a better separation of all three columns (Figure 4). However, visual review of the chromatograms for each column resulted in the selection of the Atlantis T3 based on resolutions and peak shapes.

Figure 4: AutoChrom MS combined chromatograms of the composite samples for the column screen, using a 5 – 95% acetonitrile/water gradient with 0.1%TFA at 40°C. The most optimizable column was Atlantis T3. Components labeled with a “C_mass” had low MS signals and were tracked initially by their UV spectra; in the subsequent optimization the MS signals were used.
The initial model translated into the “synthetic” chromatograms generated by AutoChrom MS (Figure 4). In Figure 4 the peaks are labeled with their m/z followed in some cases by their major fragment (e.g., 190_162). Components labeled with a “C_mass” had low MS signals and were tracked initially by their UV spectra; in the subsequent optimization the MS signals were used. After the column and mobile phase selection, the method development algorithm was based on the AutoChrom MS combined composite chromatogram that was further used for gradient and mobile phase modifier (i.e., TFA) optimization.
Once the Atlantis T3 column was selected, in addition to the initial 5-95% B (30 min) run, two additional gradients were proposed by AutoChrom MS, 19 - 95% B (51 min) and 5 – 95% B (60 min). These three gradients were run on the composite samples to collect the LCMS/ LC-UV data and they were used in the model for the gradient and %TFA optimization, optimizing start point (1D), end point (2D), time of gradient (3D), and %TFA (4D). As the model evolved with every new experimental data in an iterative way, two additional TFA concentrations were added to the model (0.05%, 0.08%) to obtain a matrix of a 3 x 3 gradient/%TFA. A 2D resolution map of TFA concentration vs. the gradient end point (%B) is presented in Figure 5. By inspection of Figure 5, where the resolution changes from high to zero to high (dark blue groves), the critical pair peaks are switching order of elution. Based on the critical resolution pair of Figure 5, the optimum TFA concentration was 8.56 mM which corresponds to 0.098% v/v TFA. Therefore, the 0.10 % TFA (8.77 mM) was considered as optimum for further gradient optimization. Although the 2D map suggested a final 98%B for the method, we subsequently discovered that this value was an overestimation (vide infra).

Figure 5: 2D resolution map “slice” for the composite sample for 3 gradients and 3 TFA concentrations. The concentration of 0.10% TFA was chosen for further gradient optimization.
In order to optimize the gradient steps (including the final %B), the best 4 injections/gradients were selected (Table 3), top 4 gradient pairs). Based on this data, quadratic models (see eq. 1) were generated for the elution time (tR) of selected 15 components where we had authentic samples to cover the whole range of hydrophobicity. From the quadratic model 3 new gradients were predicted with the aim to improve the suitability (value normalized to 1 in the software; common criteria were resolution, run time and retention factor, k’). The expectation was to increase the suitability from 0.4 of the “12-26% (6 min);…” gradient to 0.77 predicted for the “12-31% (1.42 min);…” gradient (Table 3, bottom row for each gradient).
| Gradient | Suit. | Peaks(m/z) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 206.06 | 190.07 | 162.08 | 365.18 | 318.2 | 190.18 | 393.22 | 395.23 | 421.25 | 203.12 | 503.21 | 427.15 | 485.2 | 521.18 | 485.2_1 | ||
| 14-32% (6 min); 32-33% (8 min);33-94% (9min) | 0.571 | 4.23 | 6.23 | 6.49 | 10.86 | 13.06 | 13.66 | 16.09 | 17.57 | 19.85 | 21.07 | 21.92 | 22.39 | 23.63 | 23.79 | 24.1 |
| 0 | 4.1 | 6.22 | 6.48 | 10.93 | 13.2 | 13.78 | 16.3 | 17.79 | 19.84 | 21.06 | 21.92 | 22.4 | 23.67 | 23.83 | 24.12 | |
| 15-32% (5 min); 32- 34% (21 min); 34-99% (8min) | 0 | 3.59 | 5.71 | 5.98 | 10.07 | 12.3 | 12.89 | 15.36 | 16.89 | 24.85 | 28.7 | 32.79 | 33.26 | 34.66 | 34.76 | 35.02 |
| 0 | 3.63 | 5.71 | 5.98 | 10.02 | 12.18 | 12.79 | 15.19 | 16.7 | 24.85 | 28.7 | 32.79 | 33.26 | 34.6 | 34.72 | 34.99 | |
| 12-26% (6 min); 26- 27% (12 min); 27-82% (9min) | 0.415 | 5.51 | 7.48 | 7.73 | 15.38 | 20.08 | 21.75 | 24.1 | 24.4 | 25.3 | 26.5 | 27.34 | 27.83 | 29.15 | 29.32 | 29.64 |
| 0.422 | 5.67 | 7.49 | 7.74 | 15.38 | 20.08 | 21.75 | 24.05 | 24.36 | 25.33 | 26.51 | 27.34 | 27.81 | 29.14 | 29.29 | 29.63 | |
| 6-21% (1 min); 21-23% (46 min); 23-64% (4min) | 0 | 5.4 | 6.55 | 6.84 | 23.85 | 32.87 | 37.08 | 52.56 | 52.83 | 53.52 | 54.08 | 55.12 | 55.63 | 58.02 | 58.37 | 59.37 |
| 0 | 5.38 | 6.55 | 6.84 | 23.85 | 32.86 | 37.08 | 52.56 | 52.79 | 53.46 | 54.06 | 55.13 | 55.63 | 58.01 | 58.37 | 59.37 | |
| 12-31% (1.42 min); 31-33% (7.1 min); 33- 98% | 0.765 | 4.7 | 8.69 | 7.55 | 8.04 | 10.41 | 10.96 | 12.94 | 13.42 | 14.32 | 15.49 | 16.06 | 16.52 | 17.21 | 17.33 | 17.6 |
| - | ||||||||||||||||
| 12-30% (1.42 min);30-33% (7.1 min); 33-76% | 0.746 | 4.72 | 7.69 | 7.09 | 8.42 | 10.85 | 11.43 | 13.19 | 13.59 | 14.47 | 15.7 | 16.3 | 16.76 | 17.62 | 17.77 | 18.06 |
| - | ||||||||||||||||
Table 3: Predicted tR (top row of the gradient) and suitability using the best gradients and quadratic model (top 4 row pairs).
At this time the suitability options were changed to better accommodate the model (suitable Run Time was changed to 10t0 instead of 20t0). After running the proposed gradients, the results were totally unexpected. Suitability appeared to be zero for both proposed gradients: “12-30% (1.42 min);…” and “12-31% (1.42 min);…”.
The error in prediction was mainly a modeling issue (i.e., the retention time of the components had suboptimal descriptors). In the case of the component with m/z=206 there is a systematic deviation from the straight line of the experimental tR vs. the calculated one (Figure 6). Component with m/z=206 is also the first-eluting peak in the chromatogram, and the model fails in this region of the gradient. However, for the later eluting components there is a good correlation between the experimental tR values and the predicted ones (m/z=503, Figure 6).

Figure 6: Predicted vs. experimental tR of the component at m/z=206 (left) and m/z=503 (right).
This can be explained by understanding the particularities of quadratic models; they work well for interpolation, but not for extrapolation. This limitation of the quadratic model can be seen in a plot of the %B of where a particular component elutes vs. the retention time tR of this component. By placing the 4 experimental data pairs (tR, % B) used for the modeling (Table 3) on the graph, the design space, we obtain a certain range of %B for a component. If in the subsequent gradients the %B of that component falls within the range (%B) used for the modeling, there is a good chance of a match between the predicted and experimental values (Figure 7, m/z=206 and m/z=503).

Figure 7: Predicted and experimental point for m/z=206 (left) and m/z=503 (right) in the gradient starting with 12-31% (1.42 min…). The design space (blue diamonds) represents the data points used in the quadratic model.
For components m/z=190.07 (displayed with 2 decimal place values because we have 2 components with m/z=190) and m/z=162 the discrepancy between the predicted and experimental data is significant (Figure 8). For component m/z=190.07 the %B used for the modeling was between 20% and 22%, but on the predicted chromatogram %B value for elution of the component appears to be very far from this range, close to 32% (Tables 3 and 4), gradient “12-31% (1.42 min)...”. As a result there is a significant error in the prediction of tR for this component. The difference between the experimental and predicted tR is ~3 min (Figure 8). For component m/z=162, the difference is ~2 min.
| Run | Experiment | Suit | Peaks (m/z ) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 206 | 190.07 | 162.08 | 365.18 | 318.2 | 190.18 | 393.22 | 395.23 | 421.25 | 203.12 | 503.21 | 427.15 | 485.2 | 521.18 | 485.2_1 | |||
| 1 | 5-95% (30 min) | 0 | 7.97 | 9.57 | 9.81 | 13.96 | 15.46 | 15.56 | 16.06 | 16.46 | 17.75 | 19.85 | 20.77 | 21.83 | 24.38 | 24.66 | 25.31 |
| 2 | 5-95% (40 min) | . | 8.69 | 10.69 | 10.94 | 16.6 | 18.33 | 19.41 | 19.91 | 21.62 | 23.93 | 25.35 | 26.69 | 30.06 | 30.43 | 31.24 | |
| 3 | 5-95% (60 min) | 0.364 | 9.91 | 12.6 | 12.86 | 21.5 | 23.79 | 24.26 | 25.68 | 26.41 | 28.92 | 31.43 | 33.98 | 35.8 | 40.79 | 41.36 | 42.42 |
| 4 | 14-32% (6 min); 32-33% (8 min); 33- 94% (9 min) |
0 | 4.1 | 6.22 | 6.48 | 10.93 | 13.2 | 13.78 | 16.3 | 17.79 | 19.84 | 21.06 | 21.92 | 22.4 | 23.67 | 23.82 | 24.12 |
| 5 | 15-32% (5 min); 32-34% (21 min); 34-99% (8 min) |
0.425 | 3.63 | 5.71 | 5.98 | 10.02 | 12.18 | 12.79 | 15.19 | 16.7 | 24.85 | 28.7 | 32.79 | 33.26 | 34.6 | 34.72 | 34.99 |
| 6 | 12-26% (6 min); 26-27% (12 min); 27-82% (9 min) |
0.567 | 5.67 | 7.49 | 7.74 | 15.38 | 20.08 | 21.75 | 24.05 | 24.36 | 25.33 | 26.51 | 27.34 | 27.81 | 29.14 | 29.29 | 29.63 |
| 7 | 6-21% (1 min); 21- 23% (46 min); 23- 64% (4 min) |
0 | 5.38 | 6.55 | 6.84 | 23.85 | 32.86 | 37.08 | 52.56 | 52.79 | 53.46 | 54.06 | 55.13 | 55.63 | 58.01 | 58.37 | 59.37 |
| 8 | 12-30% (1.42 min); 30- 33% (7.1 min); 33-76% (5.68 min) |
0 | 2.22 | 5.04 | 5.24 | 8.29 | 10.57 | 11.2 | 13.04 | 13.44 | 14.35 | 15.58 | 16.16 | 16.58 | 17.77 | 17.92 | 18.32 |
| 9 | 12-31% (1.42 min); 31-33% (7.1 min); 33-98% (7.81 min) |
0 | 4.66 | 5.29 | 5.43 | 8.02 | 10.27 | 10.89 | 12.85 | 13.29 | 14.18 | 15.35 | 15.89 | 16.27 | 17.27 | 17.39 | 17.65 |
| 10 | 15-33% (6 min); 33-36% (3 min); 36-73% (5 min); 73- 80% (4 min) |
0 | 3.89 | 6 | 6.28 | 10.48 | 12.08 | 12.47 | 13.43 | 13.75 | 14.56 | 15.88 | 16.39 | 16.84 | 18.12 | 18.31 | 18.72 |
| 11 | 15-23% (17 min); 23-41% (7 min); 41- 71% (3 min) |
0 | 3.86 | 6.76 | 7.18 | 23.93 | 25.52 | 25.98 | 27.28 | 27.72 | 28.6 | 29.61 | 30.17 | 30.55 | 32.03 | 32.27 | 32.81 |
| 12 | 15-23% (1.5 min); 23-34% (12 min); 34-69% (4 min) |
0.599 | 5.67 | 6.59 | 6.81 | 12.61 | 15.09 | 15.83 | 17.82 | 18.21 | 18.99 | 20.1 | 20.69 | 21.15 | 22.92 | 23.2 | 23.82 |
Table 4: Experimental results for all runs used to build the model.

Figure 8: Predicted and experimental point for m/z=190.1 (left) and m/z=162 (right) in the gradient starting with 12-31% (1.42 min…). The design space (blue diamonds) represents the data points used in the quadratic model.
Because of the deficiencies with the quadratic model discussed in the above analysis, a successful automated method development needs to include the use of linear models and a wider range of gradients. Also, the use of one step gradients leads to better results in the generation of the peak model, than multi-step gradients. Therefore, we added in the modeling 3 additional linear gradients: 5-95% (30 min), 5-95% (40 min) and 5-95% (60 min) (Table 4, first three rows). Using the already constructed peak models, 3 new injections/gradients were generated: “15-33% (6 min); ...”, “15-23% (17 min); ...” and 5-23% (1.5 min); …” (Table 4, last three rows). In addition, the optimization parameters used at the first step were changed to: Suitable Run Time =10t0 instead of 20t0 and Maximal Run Time was kept at 30t0.
With a challenging sample mixture, there was a need to increase the suitability for all injections. As a result we were able to see a maximal suitability of 0.599 for the last injection (Table 4). With all these changes, all 12 gradients listed in Table 4 were introduced into the LC Simulator and the output is listed in Table 5. By inspection of Table 5, the suitability calculations were different as compared with those in Table 4, but their values were in the same order. The last run/ gradient (#12) was found to be the best, with good resolution (Min Rs = 1.337) and a relatively short run time (24 min). Gradients 6 and 7 gave good minimum resolutions (1.349 and 1.658), but at the cost of significantly longer run times.
| Run | Suitability | Min Rs | Mean Rs | Run Time | Rs Score |
|---|---|---|---|---|---|
| 1 | 0 | 0.283 | 9.752 | 25.539 | 0.929 |
| 2 | 0.536 | 1.571 | 11.717 | 31.514 | 0.929 |
| 3 | 0.243 | 1.147 | 12.398 | 42.768 | 0.901 |
| 4 | 0 | 0.86 | 8.339 | 24.256 | 0.8 |
| 5 | 0.319 | 1.152 | 10.395 | 35.123 | 0.95 |
| 6 | 0.469 | 1.349 | 9.744 | 29.766 | 0.978 |
| 7 | 0 | 1.658 | 10.682 | 59.975 | 1 |
| 8 | 0 | 0.972 | 8.313 | 18.478 | 0.901 |
| 9 | 0 | 0.97 | 7.064 | 17.766 | 0.843 |
| 10 | 0 | 0.781 | 8.142 | 18.895 | 0.888 |
| 11 | 0 | 0.989 | 12.212 | 33.038 | 0.919 |
| 12 | 0.505 | 1.337 | 9.288 | 24.052 | 0.977 |
Table 5: The LC Simulator output for all gradient runs
The peak model is shown in Table 6. There is a good agreement between the experimental retention times (tR,exp, min) and predicted ones (tR, min). The worst asymmetry (As) is for the peak at m/z=318, a quaternary ammonium compound. Based on this model a chromatogram was predicted for gradient run 12 and was verified by an experimental run (Figure 9). There were good matches between the predicted and experimental retention times for all 15 peaks. There is a relatively higher difference (Δ=0.7 min) between the predicted and experimental retention times (tR) at the beginning and the end of the run for the first peak and the last three peaks (Table 6 and Figure 9). The peak model (log k*) in Table 6 is a function of % B and, at the retention times of those components, has suboptimal descriptors.
| # | Peak (m/z) | tR | tR,exp | k' | k* | Peak width | As | Peak Model |
|---|---|---|---|---|---|---|---|---|
| 1 | 206.06 | 5.53 | 5.67 | 2.37 | 2.33 | 0.13 | 1.03 | a = 6.7092e+1, b = -4.4526e+2, c = 0.0000 |
| 2 | 190.07 | 7.36 | 7.49 | 3.49 | 2.5 | 0.17 | 0.94 | a = 4.6522, b = -2.7022e+1, c = 2.8483e+1 |
| 3 | 162.08 | 7.62 | 7.74 | 3.64 | 2.59 | 0.18 | 0.84 | a = 4.4817, b = -2.4967e+1, c = 2.6422e+1 |
| 4 | 365.18 | 15.49 | 15.38 | 8.44 | 6.17 | 0.3 | 1.26 | a = 7.8915, b = -3.0030e+1, c = 2.2042e+1 |
| 5 | 318.2 | 19.82 | 20.08 | 11.08 | 7.55 | 0.94 | 7.26 | a = 8.8109, b = -3.5135e+1, c = 3.5155e+1 |
| 6 | 190.18 | 21.57 | 21.75 | 12.15 | 8.13 | 0.61 | 1.31 | a = 8.5750, b = -3.1789e+1, c = 2.8010e+1 |
| 7 | 393.22 | 24.22 | 24.05 | 13.77 | 11.54 | 0.23 | 2.91 | a = 1.1572e+1, b = -4.4976e+1, c = 4.3169e+1 |
| 8 | 395.23 | 24.56 | 24.36 | 13.97 | 12.75 | 0.11 | 1.15 | a = 1.0687e+1, b = -3.7496e+1, c = 3.0379e+1 |
| 9 | 421.25 | 25.33 | 25.33 | 14.45 | 13.68 | 0.08 | 1.2 | a = 5.9729, b = -6.4499, c = -1.3655e+1 |
| 10 | 203.12 | 26.51 | 26.51 | 15.16 | 13.76 | 0.12 | 1.04 | a = 5.4452, b = -7.4977, c = -3.1652 |
| 11 | 503.21 | 27.28 | 27.34 | 15.64 | 14.99 | 0.09 | 1.08 | a = 7.2098, b = -1.0946e+1, c = -2.3356 |
| 12 | 427.15 | 27.82 | 27.81 | 15.96 | 15.31 | 0.09 | 1.11 | a = 6.6067, b = -7.4324, c = -5.2592 |
| 13 | 485.2 | 29.35 | 29.14 | 16.89 | 16.23 | 0.09 | 1.09 | a = 8.0991, b = -1.1343e+1, c = 5.6269e-2 |
| 14 | 521.18 | 29.52 | 29.29 | 17 | 16.32 | 0.1 | 1.06 | a = 7.7923, b = -1.0148e+1, c = -7.4871e-1 |
| 15 | 485.2_1 | 29.87 | 29.63 | 17.22 | 16.5 | 0.11 | 1.11 | a = 7.0134, b = -7.1988, c = -2.8127 |
Table 6: Optimized Peak Model for all 15 Components.

Figure 9: Predicted (top) and experimental (bottom) chromatograms (LC-UV) of the optimized gradient. The peaks are labeled with their m/z values.
It is important to note that for a successful model, two important parameters are needed: dwell volume (VD) and dead volume (Vo). These two values were determined according to methods reported in literature [19,20]. Failure to accurately determine these two values will result in errors in building the retention model. These errors will clearly manifest for highly retained species and for peaks eluting close to the initial gradient rounding. The version of the software used does not differentiate between the mixing and non-mixing volume of the dwell volume. Besides, it also does not clarify whether the dead volume value should be corrected for extra-column volume or not. Therefore, it is recommended to eliminate peaks eluting close to the gradient rounding volume (i.e., close to the start of the gradient, the change in the gradient slope, and at the end of the gradient) where the model may show difficulty matching the theoretical and experimental results.
Potency assay method prediction
With the stability indicating method generated, a shorter potency assay method can be derived from the long stability method by using the already established peak model while relaxing some of the suitability parameters, such as the need to resolve the minor peak from each other. Using the AutoChrom software we can make relevant only the API of interest and set suitability options to resolve these peaks from any of the other impurities peaks which, for an API assay method, do not need to be resolved from each other. A shorter run time method (e.g., 10 - 15 min) can be generated by only using computational techniques. This approach of generating a (short) potency assay method from the stability indicating (long) method ensures no co-elution with the main peaks. Figure 10 shows the HPLC predicted chromatogram for the potency/assay method.

Figure 10: Predicted HPLC chromatogram for the potency/assay method using the peak model.
The combination of automation with chemometric peak tracking and software-based decision making in AutoChrom MS was able to generate a suitable stability indicating chromatographic method for an extremely challenging sample comprising of a triple API combination drug product containing potential degradants with a wide range of hydrophobicities. For the generation of the correct peak retention models equations one-step gradients with a wide range of % B should be included into modeling. Extrapolation using quadratic models can lead to significant errors in tR prediction. Modeling can be difficult for components with low retention. The composite sample concept comprising of 15 reference standards, including the parent APIs made it possible to update the project analytical control strategy late in the method development process, and update the method to resolve an additional impurity with only a few additional experiments. There were good matches between the predicted and experimental retention times. The model behind this drug substance stability indicating method can be used for the generation of an assay/potency method conditions, using the computational tool, to be used in subsequent method development for the drug product.
The helpful comments of Adrian Clarke during the review of this paper are greatly appreciated.