Advanced Techniques in Biology & Medicine
Open Access
ISSN: 2379-1764
ISSN: 2379-1764
Research Article - (2016) Volume 4, Issue 2
The ability to resolve secondary and tertiary structures, as present in their native state within the genome of RNA viruses, is an essential first step in elucidating the replication process of these viruses. The hepatitis C virus (HCV) genome is composed of a single (+) strand RNA whose replication is controlled primarily by the 3'-untranslated region (3'UTR). The 3'UTR comprises a genotype-specific variable region (VR), a poly (U/UC) repeat and a conserved 98-base sequence called the X-tail. Although structural models of the 3'UTR from in vitro biochemical analysis have been proposed, its native structure as part of the full-length viral genome existing within the intracellular environment is uncertain. As a means to map the native double-strand (ds) and single-strand (ss) RNA structures contained in the 3'UTR of the full-length genome, we performed in situ chemical tagging in combination with site-directed RT-qPCR. Results indicate that ssRNA is predominant in the VR-poly (U/UC) repeat sequences, whereas dsRNA elements are restricted to the X-tail. In situ chemical tagging also identified a unique dsRNA element located in the VR-poly (U/ UC) repeat region comprised in part of sequences located outside the 3'UTR. The results described here constitute a first report for in situ probing and detection of secondary structures present within the 3'UTR sequence of the HCV genome. The use of intracellular RNA modifying agents to demarcate dsRNA and ssRNA, in combination with PCR to amplify trace amounts of the targeted RNA, has potential application in mapping native secondary structures within complex intracellular RNAs.
Keywords: In situ, RNA secondary structure, Hepatitis C virus genome, 3'UTR, Psoralen, Dimethyl sulphate
DMS: Dimethyl Sulphate; ds: Double-Strand; HCV: Hepatitis C Virus; NMIA: N-Methylisatoic Anhydride; NS: Nonstructural; ORF: Open Reading Frame; RT-qPCR: Reverse Transcription-Quantitative Polymerase Chain Reaction; SD: Standard Deviation; SEM: Standard Error of the Mean; SL: Stem-Loop; ss: Single- Strand; SHAPE: Selective 2'-hydroxyl Acylation Analyzed by Primer Extension; UTR: Untranslated Region; VR: Variable Region
Accurate determination of secondary and tertiary RNA structures is key to understanding RNA structure-function relationships [1]. RNA is predicted to fold in a hierarchical manner with local canonical (Watson-Crick) and non-canonical base-base pairing forming secondary structures comprised of helical double-strand (ds) sequences interspersed with small intervening single-strand (ss) hairpin loops, internal bulges and junction loops composed of three or more individual RNA strands [2]. After this initial folding, ensuing tertiary contacts stabilize overall RNA configuration and allow for tight packaging of earlier-formed RNA helices [3]. Final maturation and overall RNA structure is thought to be achieved when the entire RNA molecule reaches its lowest free-energy state [4].
Two broad approaches are commonly used conjointly to resolve RNA secondary structures, namely in silico assessment of lowest freeenergy and chemical tagging of physically unpaired (and presumed single-strand) ribonucleotides [5]. While in silico modeling has proven beneficial in predicting secondary motifs in smaller RNAs (100 to 200 bases), current algorithms lack the sophistication needed to accurately predict complex or long-range tertiary base-pairings found in larger RNA molecules [6]. Chemical tagging or footprinting of unpaired nucleotides is used to enhance the predictive value of in silico models [7-10]; however, this approach too has limits. Chemical tagging, as often performed by selective 2'-hydroxyl acylation analyzed by primer extension (SHAPE), is generally constrained to in vitro studies of small RNAs which are not in their normal cellular environment [11]. SHAPE also requires picomole amounts of a uniform RNA target per experiment [12]. These measures ensure that sufficient quantities of properly folded material are available for complete tagging and enable sufficient readout sensitivity during primer extension and subsequent capillary electrophoresis. However, when in vitro chemical tagging studies are attempted for large RNAs, there is an inherent risk of conformational trapping during the folding process that may result in aberrant RNA structures and generation of artefacts such as the underor over-representation of RNA motifs [13]. An assay system that can preserve and detect the authentic ss- and dsRNA secondary structures as found in a naturally-folded, low abundance RNA would constitute a desirable advance over existing approaches.
The hepatitis C virus (HCV) RNA genome encodes an open reading frame (ORF) measuring over 9 kb in length, flanked by two small and highly-conserved 5'- and 3'-untranslated regions (UTR) [14]. Conformational changes in these HCV UTR structures are thought to act as molecular switches to transition the virus through its various replication stages [14]. The 5'UTR is 341 nucleotides in length and is predicted to fold into a complex of four domain structures and a pseudoknot. The domain structures generally consist of an extended stem-loop structure interspersed with internal bulges or intra-domain loop junctions. The 5'UTR is largely responsible for proper positioning of host cell ribosomes for translation of the HCV ORF [15]. The HCV 3'UTR is predicted to fold into a simpler tripartite structure and is comprised of a 27 to 70 base genotype-specific variable region (VR), a poly(U/UC) repeat sequence and a highly conserved 98-base sequence called the X-tail [14]. The X-tail and the adjacent 26 to 62 nucleotidelength U/UC repeat sequence constitute the minimal required elements for synthesis of the HCV (-) strand RNA intermediate in vitro [16-18]. The VR segment, while dispensable for in vitro (-) strand RNA synthesis, appears essential for optimum viral RNA replication [16,18]. From a structural perspective, lowest free-energy models, in vitro biochemical evidence and genetic mutations predict a stable 46 nucleotide terminal stem-loop (SL1) in the X-tail [17,19-21]; however, the lack of adequate biochemical tools to determine the in vivo form of the 3'UTR has made accurate determination of the native 3'UTR structure challenging [20,22].
An examination of secondary structures present in the HCV 3'UTR, as they exist in their native intracellular state, will enable a more rational assessment of the contribution of the HCV 3'UTR to viral replication. As a means to detect native dsRNA and ssRNA elements in a low abundance intracellular RNA species, we performed in situ chemical crosslinking and nucleotide tagging, in combination with sitedirected RT-qPCR, to detect and demarcate dsRNA and ssRNA motifs contained within the 3'UTR sequence of the intracellular HCV genome. Th e results described here constitute a first report for in situ probing and detection of secondary structures present within the 3'UTR sequence of the HCV genome.
Generation of HCV RNAs
Full-length HCV 1b genomic RNA was generated from the HCV RNA expression plasmid (p)ER-1b. pER-1b is a derivative of pGEM-7Zf- HCV1b with addition of the microRNA-122 complementary sequence and the HCV 1b 3'UTR (cDNA template RNA for HCV 1b was kindly provided by Dr Hugo Soudeyns, CHU Sainte-Justine, Montreal) [23,24]. Changes were introduced into the pGEM T7 RNA polymerase promoter GAATACAAGCTTGTAATACGACTCACTATAGCCAGCCCCC to enable RNA transcription to start at the authentic HCV genome terminal guanine nucleotide (underlined). pER-1b was altered by site-directed mutagenesis to contain the NS5b RNA polymerase GDD→GND mutation [25] or to include an additional frame-shift mutation within the leftward end of the NS2 coding sequence (see Table 1 for primer sequences and corresponding HCV genome positions). The NS5b GND mutation was expected to maintain intracellular HCV RNA in a pre-replicative state by preventing viral polymerase synthesis of the (-) strand RNA replication intermediate and hence arresting subsequent viral replication [26]. Th e NS2 frame-shift mutation gives rise to a non-functional truncated NS2 protein and a complete loss of viral replication proteins NS3, NS4a/b and NS5a/b. The predicted DNA sequences for pER-1b, pER-1b GND mutant and pER-1b NS2 mutant constructs were verified by DNA sequencing.
| Probe 1-9380-9404-Sense (S) | GAG CTA AAC ACT CCA GGC CAA TAG G |
| Probe 1-9588-9607-Antisense (AS) | ATG ATC TGC AGA GAG GCC AG |
| Probe 2-9380-9404-S | GAG CTA AAC ACT CCA GGC CAA TAG G |
| Probe 2-9546-9565-AS | GCG GCT CAC GGA CCT TTC AC |
| Probe 3-9509-9528-S | TTT GGT GGC TCC ATC TTA GC |
| Probe 3-9588-9607-AS | ATG ATC TGC AGA GAG GCC AG |
| Probe 4-9380-9404-S | GAG CTA AAC ACT CCA GGC CAA TAG G |
| Probe 4-9509-9528-AS | GCT AAG ATG GAG CCA CCAAA |
| 1GND mutant 8541-8561 | GTT AAC GGA AAC GAC CTC GTC |
| 2NS2 mutant 3025/3026 | CCA TTC TAA TTG CCA TAC TCG GTC C |
1GDD→GND mutation G/A that abolished NS5b RNA polymerase activity [25].
2NS2 reading frame shift mutant resulting from removal of nucleotides TA (strikethrough).
Table 1: Probe oligonucleotide sequences.
HCV wild-type and mutated genomic RNAs were initially synthesized with the MEGAscript T7 kit (Life Technologies, AM1333) and 1 μg of XbaI-linearized (Life Technologies, 15226012) mung nuclease-treated (New England BioLabs, M0250) plasmid DNA [27,28]. Various HCV 3'UTR RNA fragments were also synthesized using the MEGAscript T7 polymerase following an initial pfx (Life Technologies, 11708013) high-fidelity PCR amplification step with the HCV 3'UTR DNA and 3'UTR-specific primers (Table 1). RNA transcription reactions proceeded for 4 to 6 h at 37oC, followed by DNA template removal with RNase-free DNase (Life Technologies, AM2222), organic extraction (Molecular Research Center Inc., TR118) and ethanol precipitation. Genomic RNA and 3'UTR RNAs were verified for predicted size and product integrity by resolution in 0.8% to 1% formaldehyde-agarose gels [29].
Cell culture and folding of intracellular HCV RNA templates
A volume containing 10 μg HCV genomic RNA or the 3'UTR RNA fragment was single-pulse-injected (270 V and 960 μFD, BioRad Life Science, Gene Pulser) into 4×106 Huh-7 cells (the Huh-7 human hepatoma cell line was provided by Dr Tatsuo Takahashi, Health Science Research Resources Bank, Japan) [28,30]. Following electroporation, Huh-7 cells were placed in 8 ml Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, 11995081) supplemented with 10% FBS and antibiotics, and cultured for 6 to 12 h such that viable cells could attach to the plate. This culture period was expected to allow the injected HCV RNA sufficient time to synthesize a complete complement of viral proteins, and enable its association with viral replication proteins as well as appropriate cellular RNA binding proteins (Supplementary Figure 1) [31,32]. Electroporation-damaged and non-viable unattached cells were removed and the remaining attached cells were trypsinized, pelleted through 5 ml FBS and washed three times with PBS to remove any residual extracellular HCV RNA. Cells were divided into aliquots of 1×106 cells/100 μl in 10 mM Tris-saline buffer (pH 7.5) in preparation for in situ RNA crosslinking with aminomethyl trioxsalen (psoralen, Sigma-Aldrich, P8399) or A/C ribonucleotide modification with dimethyl sulfate (DMS, Sigma-Aldrich, D186309).
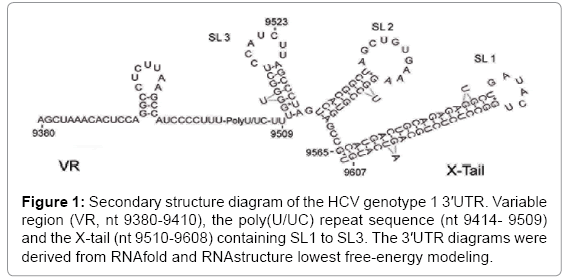
Figure 1: Secondary structure diagram of the HCV genotype 1 3′UTR. Variable region (VR, nt 9380-9410), the poly(U/UC) repeat sequence (nt 9414- 9509) and the X-tail (nt 9510-9608) containing SL1 to SL3. The 3′UTR diagrams were derived from RNAfold and RNAstructure lowest free-energy modeling.
Virus replication potential by pER-1b, pER-1b GND mutant and pER-1b NS mutant was assessed by the fluorescence-forming unit (ffu) assay [28,30]. This assay enables detection of viral structural proteins, represented by core protein (Santa Cruz Biotechnology, anti-HCV core proteins, sc-52403), and non-structural proteins (NS), represented by NS5b protein (Santa Cruz Biotechnology, anti-NS5b monoclonal antibody, sc-58146). All three HCV constructs were capable of synthesizing their particular complement of viral proteins by 12 h posttransfection (Supplementary Figure 1); however, only wild-type ER-1b RNA was capable of completing an entire replication cycle to produce infectious virions (~3×104 infectious units (IU)/ml).
In vitro and in situ crosslinking of dsRNA
Psoralen was chosen as a means to localize dsRNA elements in the HCV 3'UTR. This was based on the known ability of psoralen to freely penetrate living cells and form chemical crosslinks in dsRNA, while preserving native RNA structures and RNA interactions with cognate binding proteins [33].
In vitro crosslinking by psoralen was performed after resuspending 4 μg of 3'UTR RNA (HCV 1b nt 9380-9609) in 20 μl RNA folding buffer (25 mM Tris-HCl, 200 mM NaCl, 10 mM MgCl2 and 0.1 mM EDTA, pH 7.5), heating the RNA to 95°C for 5 min and allowing RNA folding in a two-series slow-cooling step. The first slow-cooling step consisted of a 5°C/2 min cooling and a 10 min stationary step at 65°C. The second slow-cooling step consisted of a 2.5°C/min cooling to a final temperature of 37°C. This protocol was expected to facilitate and maintain proper folding of the 3'UTR RNA into a homogeneous population of lowest free-energy structures [34]. After the RNA was renatured, freshly-prepared psoralen was added to a final concentration of 100 μg/ml, and the samples were left to incubate at 37°C in the dark for 10 min prior to UV crosslinking.
In situ crosslinking by psoralen was performed on Huh-7 cells electroporated with the 3'UTR fragment or with the full-length HCV genome. The cells were seeded as 50 μl droplets on paraffin and placed on an ice bed. Cells were combined with psoralen (500 μg/ml final concentration) and incubated in the dark for 10 min to permit cell penetration and intercalation of psoralen into dsRNA in preparation for UV crosslinking [33].
In vitro and in situ RNA was crosslinked by irradiation with longwave (365 nm) UV light for 30 min (Spectronics, Spectrolinker XL- 1000 UV crosslinker). RNA fragments and live cells were irradiated at a distance of 9 cm from the light source for an expected energy yield of 4000 μJ/cm2.
In experiments to validate psoralen’s specific recognition and crosslinking of dsRNA sequences but not ssRNA sequences, 3'UTR X-tail RNA, which is predicted to fold almost entirely into dsRNA (Figure 1), was heated to 95°C for 5 min and slow-cooled to 70°C. X-tail RNA was then maintained at 70°C throughout the entire crosslinking procedure. This was expected to prevent dsRNA annealing of the X-tail helices [17,21]. Following TRI-reagent organic extraction and ethanol precipitation to remove free psoralen, the RNA was resuspended at 1 μg/μl in RNase-free water and stored at -80°C.
In vitro and in situ modification of ssRNA by dimethyl sulphate
DMS was chosen over the more commonly used N-methylisatoic anhydride (NMIA) to best localize ssRNA elements in the HCV 3'UTR. This was based on the reported ability of DMS to readily penetrate living cells relative to NMIA [12]. Although NMIA is capable of tagging all four ribonucleotides, DMS possesses a comparable or superior accuracy in recognizing unpaired A/C ribonucleotides [35].
In vitro DMS modification of ssRNA was performed after resuspending 4 μg of the 3'UTR RNA fragment in 20 ul of RNA folding buffer, heating the RNA to 95°C for 5 min and refolding the RNA in the slow-cooling step procedure as outlined above. Fresh DMS solution was added to the RNA to achieve a final concentration of 105 mM, after which the RNA samples were incubated at 37°C for 30 min.
In situ RNA modification by DMS was performed on electroporated Huh-7 cells suspended in 100 μl PBS to which DMS was added to a final concentration of 420 mM [36]. Cells were incubated for 10 min with continuous mixing. In vitro and in situ DMS reactions were stopped by addition of 5 μl β-mercaptoethanol.
In experiments to validate DMS recognition of ssRNA sequences but not dsRNA sequences, the 3'UTR X-tail was heated to 95°C for 5 min and slow-cooled to 70°C. X-tail RNA was then maintained at 70°C throughout the entire DMS tagging procedure to prevent dsRNA annealing and allow free access of DMS to exposed A/C ribonucleotides.
Organic extraction and ethanol precipitation were used to remove unbound DMS and β-mercaptoethanol. Treated RNA was then resuspended at 1 μg/μl in RNase-free water and stored at -80°C.
Detection of ssRNA and dsRNA
Psoralen-crosslinked or DMS-modified RNA, in conjunction with a library of primers demarcating specific structural elements in the HCV 3'UTR (Table 1 and Figure 1), were used to generate radiolabeled cDNA probes by RT-PCR. Both psoralen crosslinking and DMS modification are expected to block reverse transcriptase, thereby yielding abortive cDNA transcripts and leading to the effective loss of radiolabeled cDNA probe [35-37]. 32P-radiolabeled cDNA probes were generated from 40 ng of in vitro crosslinked/modified RNA or 500 ng of in situ crosslinked/ modified RNA. Probes were synthesized in 25 μl of QuantiTect reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) kit buffer (Qiagen, 210212) containing 0.5 μM of sense and antisense primers and spiked with 2 μl of [α32P]-dCTP (800 Ci/mmol, 10 mCi/ ml; Perkin-Elmer, Woodbridge, BLU013). The exact PCR cycle which was most reflective of psoralen crosslinking or DMS modification of 3'UTR RNA and, therefore, most efficient for future probe generation conditions was determined for each RNA sample from an initial RTqPCR screening and the use of Probe 1 primers. Probe 1 primers encompass the entire 3'UTR region. Probe working conditions were assayed in the Stratagene Mx3000P real-time PCR unit (SABiosciences). For each probe set, individual radiolabeled probes were generated from untreated (UT) RNA, RNA exposed to psoralen (P), RNA exposed to UV (UV), RNA exposed to both psoralen and UV (P+UV) or RNA modified with DMS. Individual probes were hybridized to Southern blots containing a 270 bp DNA fragment which encompassed the entire HCV 1b 3'UTR [38]. Following a high stringency wash, blots were exposed to X-ray film and changes in band intensity were measured using MacBiophotonics Image J software (Rasband, W.S., U.S. National Institutes of Health, Bethesda, Maryland, USA). Signal image units (IU) were based on 0.5×1 cm box-scans for individual blot bands and the IU were calculated as the number of pixels/0.5×1 cm box, after subtraction of background pixels/0.5×1 cm box scanned from an autoradiograph area located outside blotted DNA gel lanes.
Northern blot analysis of HCV X-tail dsRNA
To resolve whether psoralen or DMS altered PCR primer access or annealing to the HCV RNA template during PCR probe generation, 4 μg of refolded 3'UTR RNA fragment was exposed to 100 μg/ml psoralen and UV, or to 105 mM DMS. The modified RNA was denatured in a formaldehyde-agarose gel, blotted onto nitrocellulose and hybridized overnight at 50°C with 32P-radiolabeled antisense oligonucleotides for Probes 1 and 3 (Table 1) [39]. Autoradiographs were digitally scanned and changes in psoralen- or DMS-treated RNA band intensity were then compared to unmodified RNA.
Quantification of HCV RNA
Quantification of HCV RNA was performed as outlined by Takeuchi et al. [40] using the QuantiTect SYBR Green RT-PCR Kit (Qiagen, 204243) and universally conserved 5'UTR primer set CGGGAGAGCCATAGTGG (sense, genome position 130-146) and AGTACCACAAGGCCTTTCG (anti-sense, genome position 290- 272). Log10 dilutions of synthetic HCV ER-1b RNA ranging from 106 to 102 copies served as reference standards.
RNA computer modeling and molecular or statistical analysis
HCV 3'UTR secondary structure predictions were performed using RNAfold (http://rna.tbi.univie.ac.at) in conjunction with RNA structure (http://rna.urmc.rochester.edu/RNAstructureWeb) [41,42]. RNA structures were graphed using the RNA visualization tool jViz. Rna 2.0 (http://jviz.cs.sfu.ca/index.html) [43]. Selection of PCR primers and mutated HCV sequences was aided by the use of Vector NTI 8.0 software (Informax) [44]. Statistical analysis was performed using Excel (Microsoft Inc., Microsoft Office 2007).
Validation of 3'UTR dsRNA and ssRNA detection reagents
Consensus prediction from RNAfold and RNAstructure algorithms indicates that the HCV ER-1b 3'UTR VR forms a small four-G:C-pair stem. No dsRNA structures are predicted for the poly (U/UC) repeat, while the X-tail is predicted to contain almost exclusively dsRNA in the form of three dsRNA stem-loop structures (Figure 1). In respect of convention, the X-tail stem-loop structures were designated as SL1 (nt 9565-9608), SL2 (nt 9535-9561) and SL3 (nt 9511-9532) [45].
Prior to the examination of ssRNA and dsRNA elements present de novo in the HCV 3'UTR, we characterized the specificity and optimal working conditions for the dsRNA recognition reagent psoralen and for the ssRNA recognition reagent dimethyl sulphate (DMS) and validated their individual mechanism of action [46,47]. HCV RNA was exposed in vitro to increasing concentrations of psoralen, or to a fixed amount (105 mM) of DMS with increased incubation times [36]. As shown in Figure 2A and 2C, increasing the amount of psoralen or raising DMS incubation times resulted in a log10 or greater reduction in the number of detectable HCV RNA copies when measured by RT-qPCR. From these in vitro experiments we established the working psoralen concentration and the incubation time for DMS as 340 nM and 30 min, respectively. Under these conditions we observed an average delay of three qPCR cycles for the 3'UTR X-tail RNA treated with either psoralen or DMS (Figures 2B and 2D). Since a one-cycle delay represents an approximate three-fold loss in PCR signal [48], these elected conditions were expected to generate quantifiable differences in probe labelling and subsequent probe signal intensity (Figure 2).
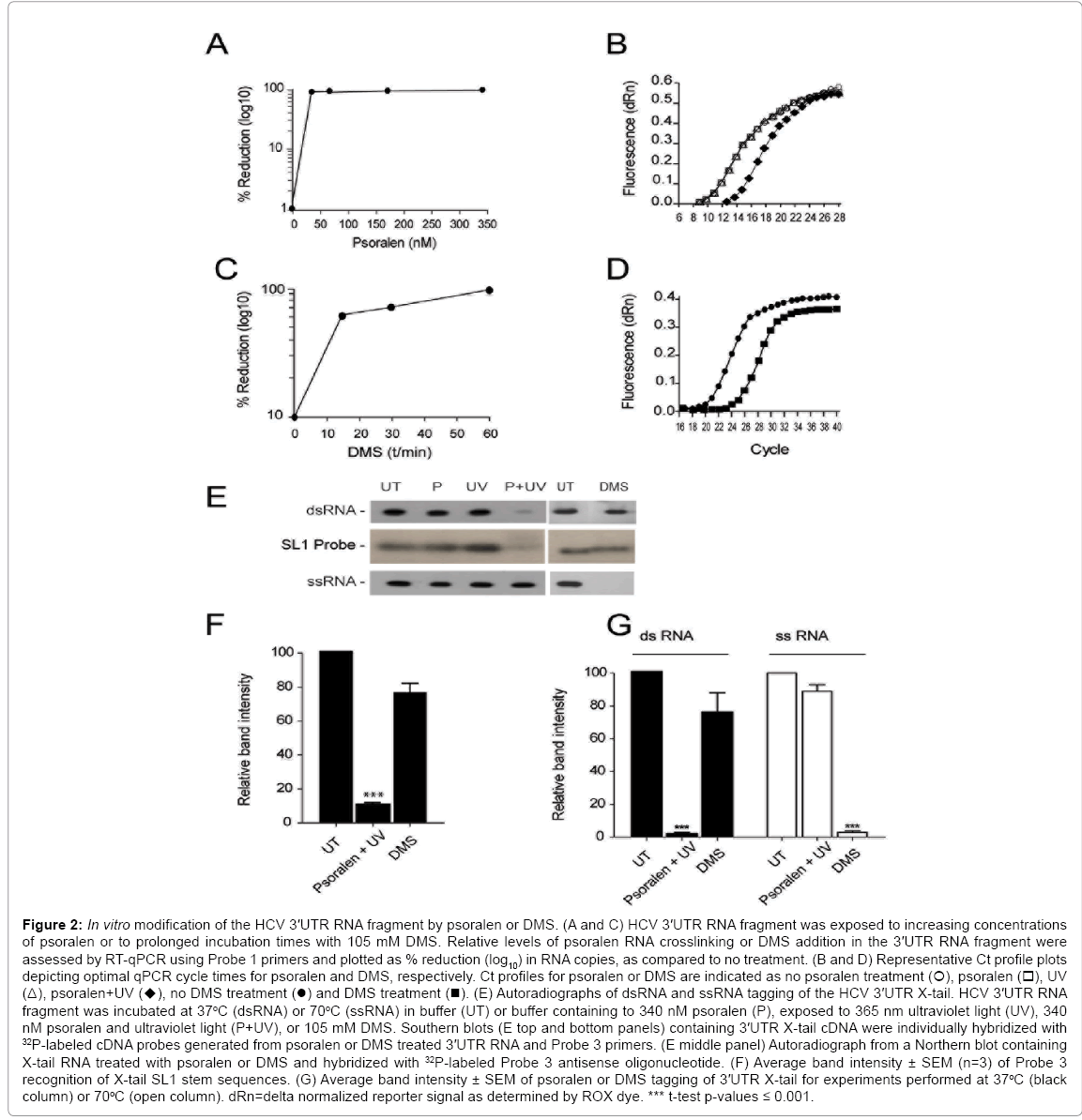
Figure 2: In vitro modification of the HCV 3′UTR RNA fragment by psoralen or DMS. (A and C) HCV 3′UTR RNA fragment was exposed to increasing concentrations of psoralen or to prolonged incubation times with 105 mM DMS. Relative levels of psoralen RNA crosslinking or DMS addition in the 3′UTR RNA fragment were assessed by RT-qPCR using Probe 1 primers and plotted as % reduction (log10) in RNA copies, as compared to no treatment. (B and D) Representative Ct profile plots depicting optimal qPCR cycle times for psoralen and DMS, respectively. Ct profiles for psoralen or DMS are indicated as no psoralen treatment (), psoralen (△), UV (Δ), psoralen+UV (♦), no DMS treatment (●) and DMS treatment (■). (E) Autoradiographs of dsRNA and ssRNA tagging of the HCV 3′UTR X-tail. HCV 3′UTR RNA fragment was incubated at 37oC (dsRNA) or 70°C (ssRNA) in buffer (UT) or buffer containing to 340 nM psoralen (P), exposed to 365 nm ultraviolet light (UV), 340 nM psoralen and ultraviolet light (P+UV), or 105 mM DMS. Southern blots (E top and bottom panels) containing 3′UTR X-tail cDNA were individually hybridized with 32P-labeled cDNA probes generated from psoralen or DMS treated 3′UTR RNA and Probe 3 primers. (E middle panel) Autoradiograph from a Northern blot containing X-tail RNA treated with psoralen or DMS and hybridized with 32P-labeled Probe 3 antisense oligonucleotide. (F) Average band intensity ± SEM (n=3) of Probe 3 recognition of X-tail SL1 stem sequences. (G) Average band intensity ± SEM of psoralen or DMS tagging of 3′UTR X-tail for experiments performed at 37oC (black column) or 70oC (open column). dRn=delta normalized reporter signal as determined by ROX dye. *** t-test p-values ≤ 0.001.
When the X-tail RNA fragment was psoralen-crosslinked at 37°C, we observed a 95% loss in signal intensity as compared to untreated RNA (Figure 2E, dsRNA, UT versus P+UV). The 95% decrease in X-tail probe (Probe 3) signal intensity following psoralen-crosslinking contrasted with the high intensity signal of X-tail RNA exposed only to UV or psoralen (Figure 2E, dsRNA, 37°C, UT versus P or UV). Compared to untreated RNA, X-tail RNA exposure to either psoralen or UV showed reductions of only 11% and 30%, respectively.
In complement and contrast to psoralen, the high percentage of dsRNA in the X-tail precluded DMS reactivity [35]. As shown in Figure 2E, dsRNA, X-tail RNA incubated at 37°C and treated with DMS showed no significant reduction in Probe 3 radiolabeling and yielded band signal intensity similar to untreated X-tail RNA (Figure 2E, dsRNA, 37°C, UT versus DMS).
In order to validate the observed targeting of psoralen to dsRNA and of DMS reactivity with (unpaired) ssRNA, refolded 3'UTR RNA fragment was exposed to psoralen+UV or to DMS prior to denaturation by formaldehyde and hybridization with a radiolabeled oligonucleotide that specifically recognizes the stem element in X-tail SL1 RNA sequences. Since SL1 has been shown to form a stable stem-loop structure in vitro [19,20], psoralen crosslinking of 3'UTR RNA prior to formaldehyde denaturation was predicted to prevent probe annealing and to reduce band intensity. It was also predicted that SL1 sequences would be insensitive to DMS treatment and allow probe annealing. As shown in Figure 2E (SL1 probe, P+UV) psoralen crosslinking of the 3'UTR RNA fragment significantly reduced band intensity. The data gathered from three experiments indicate an average loss of 89% ± 1.6% (SEM) in band intensity when compared to the untreated 3'UTR RNA fragment (Figure 2F, Psoralen+UV). Conversely, when the 3'UTR RNA fragment was treated with DMS prior to formaldehyde denaturation, we observed an average loss of only 24% ± 6% (n=3) in band intensity as compared to untreated 3'UTR RNA (Figure 2E, SL1 DMS and Figure 2F, DMS). These results indicate that observed changes in band signal intensity during PCR probe generation were likely due to prevention of primer seeding by psoralen and/or subsequent blocking of DNA strand extension. The results also indicate that DMS permits primer annealing but prevents DNA extension during probe generation.
In order to further validate the mechanism of action for DMS as reacting specifically with the unpaired nucleotides in the ssRNA sequences, and to further exclude the possibility that psoralen and DMS improperly adduct to ssRNA and dsRNA, respectively, X-tail RNA fragment was psoralen-crosslinked or DMS-treated at 70°C. This treatment was expected to prevent dsRNA annealing within the X-tail and force the X-tail to maintain an ssRNA form. As shown in Figure 2E (ssRNA), prevention of X-tail dsRNA formation during psoralen crosslinking resulted in only a 10% decrease in band signal intensity compared to untreated X-tail RNA (Figure 2E, ssRNA, UT versus P+UV). Conversely, X-tail RNA treated with DMS at a temperature of 70°C allowed DMS tagging and resulted in a marked loss of band signal intensity (Figure 2E, ssRNA, UT versus DMS).
Three additional experiments revealed that X-tail RNA treated with psoralen and maintained at 37°C versus 70°C resulted in 98% ± 1.4% loss in band signal intensity or only 11% ± 5.7% loss, respectively, as compared to untreated RNA (Figure 2G, UT versus psoralen+UV). Conversely, X-tail RNA treated with DMS at 37°C or 70°C resulted in 24.5% ± 17.7% loss in signal intensity or 97% ± 1.4% loss, respectively, as compared to untreated RNA (Figure 2G, UT versus DMS).
These experiments indicated that psoralen and DMS, when used in vitro and under biological temperatures, can specifically and differentially recognize dsRNA and ssRNA. We also noted that the average difference in band intensity for untreated RNA versus psoralenor DMS-treated RNA was 32.8 image units (IU)+1.82 SD (n=6). From this statistic, it was determined that a reduction in signal intensity of ≥ 60% would exceed the lower limit at the 95% confidence level and allow for an accurate identification of dsRNA or ssRNA.
In vitro mapping of dsRNA structures contained within the 3'UTR
Hybridization of a DNA fragment representing the entire 3'UTR sequence with a library of cDNA radiolabeled probes generated after in vitro psoralen treatment and encompassing the entire 3'UTR sequence (Probe 1), the 3'UTR sequence lacking the SL1 sequence (Probe 2), sequences representing only the X-tail (Probe 3) or sequences containing only the VR and poly(U/UC) repeat (Probe 4), showed a 96% reduction in band signal intensity for probes 1 to 3 (Figure 3, left panels, UT versus P+UV). No statistical loss (28%) in signal intensity was observed using probe 4 (Figure 3A, left panels, UT versus P+UV) (Figure 3). These results indicate that dsRNA elements were present uniquely in the X-tail sequence. No dsRNA sequences were evident in the VR-poly(U/UC) repeat sequence. Results for probe 2 also indicated that, beyond the stable terminal SL1 structure, psoralen crosslinking also detected dsRNA within the SL2-SL3 region. The inability to detect the G:C stem located in the VR may be due to its instability at 37°C as the VR G:C stem has a predicted melting temperature of 32°C.
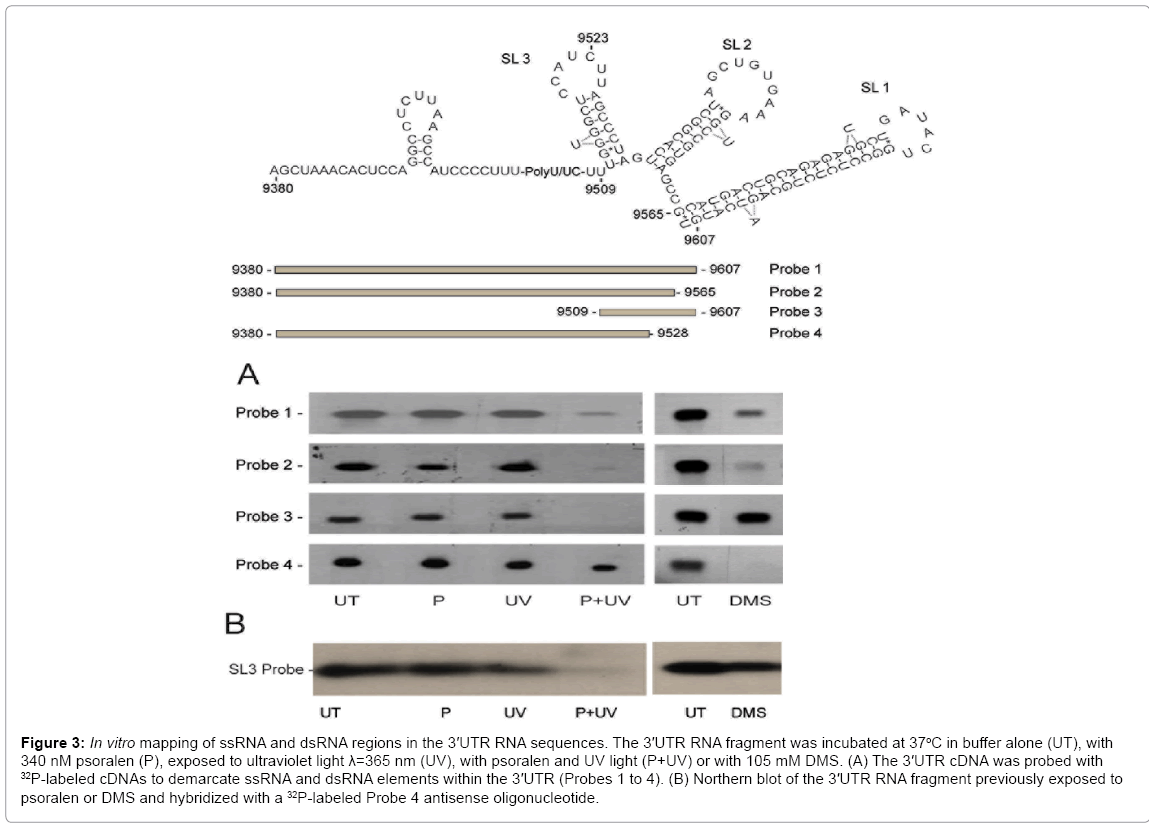
Figure 3: In vitro mapping of ssRNA and dsRNA regions in the 3′UTR RNA sequences. The 3′UTR RNA fragment was incubated at 37oC in buffer alone (UT), with 340 nM psoralen (P), exposed to ultraviolet light λ=365 nm (UV), with psoralen and UV light (P+UV) or with 105 mM DMS. (A) The 3′UTR cDNA was probed with 32P-labeled cDNAs to demarcate ssRNA and dsRNA elements within the 3′UTR (Probes 1 to 4). (B) Northern blot of the 3′UTR RNA fragment previously exposed to psoralen or DMS and hybridized with a 32P-labeled Probe 4 antisense oligonucleotide.
DMS modification recognized the poly (U/UC) repeat sequence but did not react with the X-tail (Figure 3A, right panels, Probes 1, 2 and 4 versus Probe 3). This observation was expected because computer models had predicted the X-tail to be comprised almost exclusively of dsRNA, while no dsRNA was predicted for the poly (U/UC) repeat sequences. These results further validate the capacity of psoralen and DMS to differentially recognize dsRNA and ssRNA sequences, respectively.
Probe 3 sense primer and Probe 4 antisense primer overlap in their recognition of SL3 stem sequences in the 3'UTR X-tail. These probes were also used to identify dsRNA and ssRNA, respectively, in the X-tail and the poly (U/UC) repeat (Figure 3, Probe 3 and Probe 4). We therefore examined whether X-tail SL3 formed a stable stemloop structure in vitro as depicted in Figure 1, or whether it adopted a DMS-sensitive open structure [20,22]. Refolded 3'UTR RNA fragment was treated with psoralen or DMS, denatured by formaldehyde and subsequently hybridized with Probe 4 antisense oligonucleotide. As revealed by the loss in band intensity (Figure 3B, P+UV), psoralen was able to block Probe 4 antisense primer hybridization to SL3 RNA sequences, while DMS treatment failed to significantly inhibit Probe 4 antisense hybridization (Figure 3B, UT versus DMS). The data obtained from three additional experiments indicate that psoralen reduced Probe 4 hybridization to X-tail SL3 and signal band intensity by 88% ± 2%, while DMS reduced Probe 4 antisense hybridization and signal band intensity by 20% ± 10%. These results demonstrate that the in vitro form of the X-tail SL3 is principally dsRNA.
In situ detection of dsRNA and ssRNA structures contained within the HCV 3'UTR RNA fragment
Compared to in vitro conditions, the normal concentrations of 3'UTR dsRNA or ssRNA targets for intracellular HCV genomic RNA are expected to be orders of magnitude lower than those of competing cellular dsRNA and ssRNA species [49,50]. We, therefore, established optimal working conditions for psoralen and DMS to crosslink dsRNA or mark ssRNA sequences, respectively, within the 3'UTR, as they exist in situ within the HCV genome. Addition of increased amounts of psoralen or DMS to 106 Huh-7 cells containing transfected HCV genomic RNA to levels greater than 1.7 mM and 420 mM, respectively, resulted in 99% and 84% reductions in detectable HCV genome copies as measured by RT-qPCR (Figure 4, graphs A and C). Under these conditions, we consistently observed an average delay of three PCR cycles following psoralen treatment and two PCR cycles following DMS treatment (Figure 4, graphs B and D).
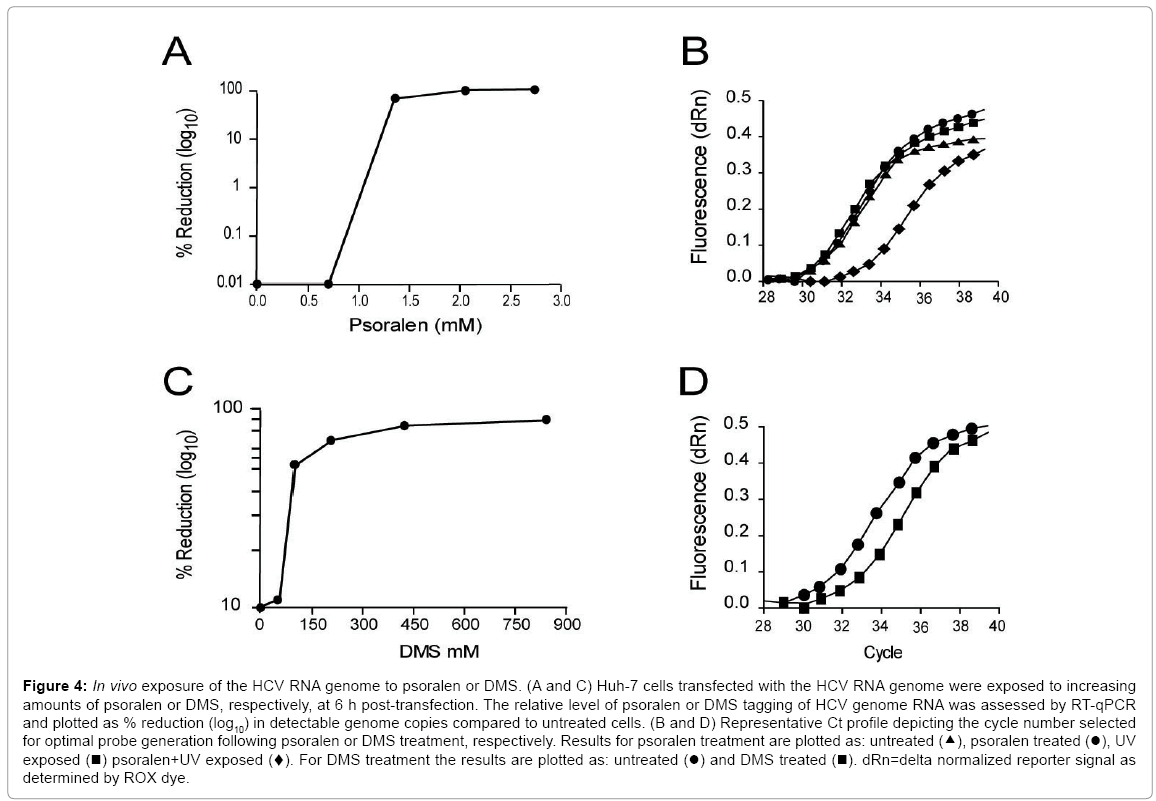
Figure 4: In vivo exposure of the HCV RNA genome to psoralen or DMS. (A and C) Huh-7 cells transfected with the HCV RNA genome were exposed to increasing amounts of psoralen or DMS, respectively, at 6 h post-transfection. The relative level of psoralen or DMS tagging of HCV genome RNA was assessed by RT-qPCR and plotted as % reduction (log10) in detectable genome copies compared to untreated cells. (B and D) Representative Ct profile depicting the cycle number selected for optimal probe generation following psoralen or DMS treatment, respectively. Results for psoralen treatment are plotted as: untreated (▴), psoralen treated (●), UV exposed (■) psoralen+UV exposed (♦). For DMS treatment the results are plotted as: untreated (●) and DMS treated (■). dRn=delta normalized reporter signal as determined by ROX dye.
In its native setting the HCV 3'UTR fragment would be expected to mimic RNA structures naturally found in the host cell RNA and to bind virus-essential cellular RNA binding proteins [51]. In order to determine whether the dsRNA and ssRNA elements observed under in vitro treatment conditions, and devoid of RNA binding proteins, were impacted by cellular RNA binding proteins [52], parallel experiments were performed in which the 3'UTR RNA fragment was psoralencrosslinked or DMS-modified in vitro (Figure 5, panels A and C), or was first electroporated into cells and given sufficient time to complex with cellular RNA binding proteins prior to in situ psoralen crosslinking or DMS modification (Figure 5, panels B and D). Results from in vitro and in situ modified HCV 3'UTR RNA revealed a significant loss in signal intensity for Probes 1 to 3 (>96%) in cells treated with psoralen (Figure 5, panels A to D) and a small loss in signal intensity for Probe 4 (less than 26%) (Figure 5, panels A and C versus panels B and D), suggesting that in situ the dsRNA elements are located in the X-tail within both the SL1 sequence and sequences comprising SL2 and SL3. The lack of significant change in signal intensity for Probe 4 suggests that the in situ form of the poly(U/UC) repeat sequence contained no detectable dsRNA (Figure 5, Probe 4). The data obtained for the in vitro and in situ DMS-modified 3'UTR fragment complemented the psoralen results by revealing that the poly(U/UC) repeat RNA sequence was susceptible to DMS modification (70% to 89% reduction for DMS, Probes 1, 2 and 4; Figure 5, panels A to D), whereas X-tail sequences remained resistant to DMS modification (Probe 3, less than 22% reduction in signal; Figure 5, panels A to D). These results indicate that the inherent folding properties of the 3'UTR fragment are not significantly altered by the presence of host cell RNA binding proteins.
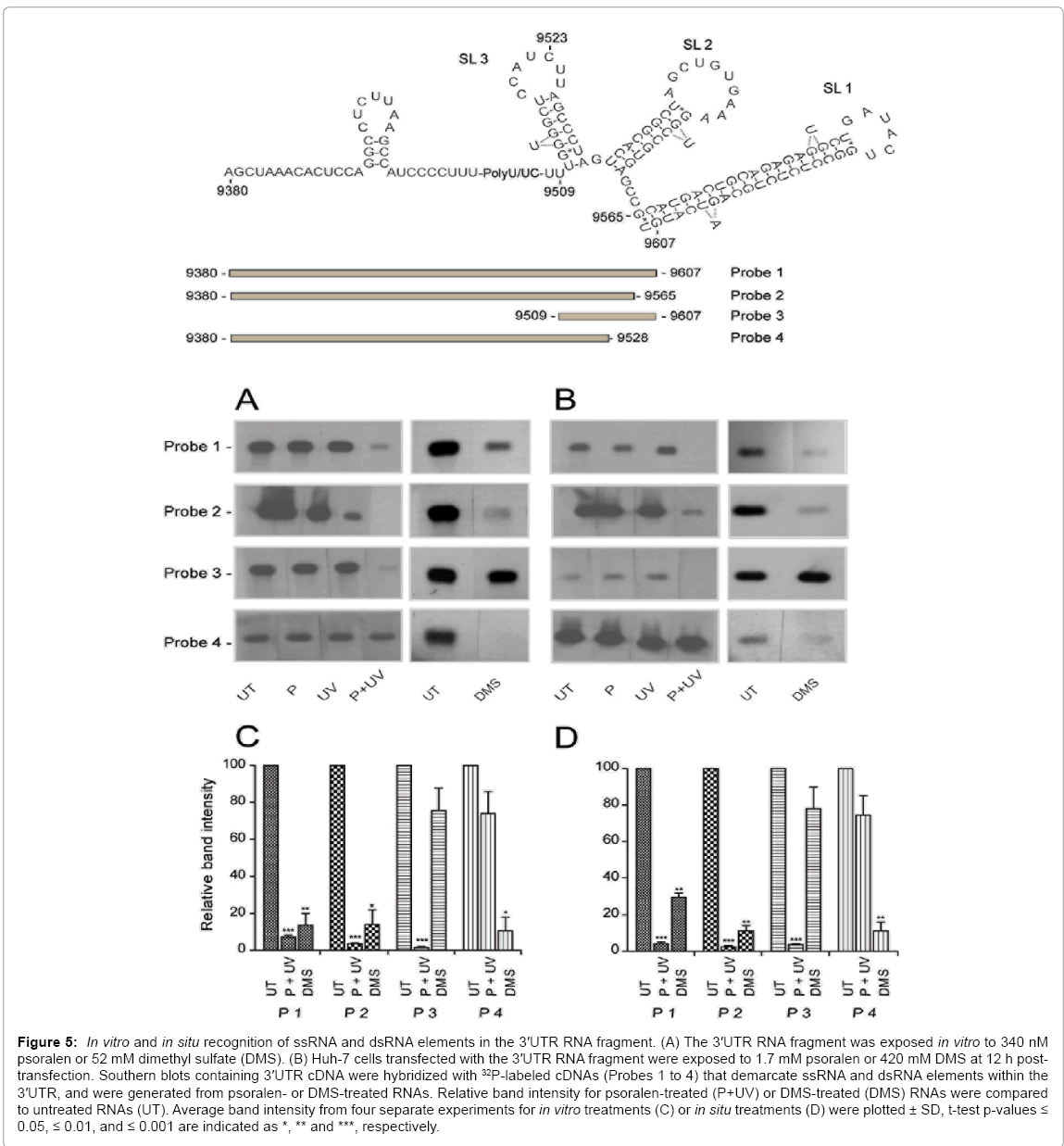
Figure 5: In vitro and in situ recognition of ssRNA and dsRNA elements in the 3′UTR RNA fragment. (A) The 3′UTR RNA fragment was exposed in vitro to 340 nM psoralen or 52 mM dimethyl sulfate (DMS). (B) Huh-7 cells transfected with the 3′UTR RNA fragment were exposed to 1.7 mM psoralen or 420 mM DMS at 12 h posttransfection. Southern blots containing 3′UTR cDNA were hybridized with 32P-labeled cDNAs (Probes 1 to 4) that demarcate ssRNA and dsRNA elements within the 3′UTR, and were generated from psoralen- or DMS-treated RNAs. Relative band intensity for psoralen-treated (P+UV) or DMS-treated (DMS) RNAs were compared to untreated RNAs (UT). Average band intensity from four separate experiments for in vitro treatments (C) or in situ treatments (D) were plotted ± SD, t-test p-values ≤ 0.05, ≤ 0.01, and ≤ 0.001 are indicated as *, ** and ***, respectively.
In situ detection of dsRNA and ssRNA structures contained within the 3'UTR sequence of the HCV genome
Although the in vitro and in situ results for the 3’UTR RNA fragment were similar and compared favorably with prior studies that employed 3'UTR fragments, the use of subgenomic fragments does not preclude, or allow detection of, more distant secondary interactions which might occur between the 3'UTR sequence and its neighboring NS5b sequence, or with sequences located distally in the 5'UTR [53-56]. In situ psoralen treatment of the entire HCV genome at 8-12 h posttransfection yielded ≥ 89% reduction in band intensity for Probes 1, 2 and 4, but showed only 27% reduction for Probe 3 (Figure 6). Further, psoralen treatment of the in situ HCV genome revealed a unique dsRNA element within the VR-poly(U/UC) repeat (Figure 6, Probe 4), which was not observed when the 3'UTR fragment was used alone (Figure 5B, Probe 4). By contrast, in situ treatment of the HCV genome with DMS complemented our findings only for the 3'UTR fragment (Figure 5). DMS treatment still showed that ssRNA elements dominate the poly(U/ UC) repeat (Figure 6, Probe 1, 2 and 4), whereas they are not apparent in the X-tail (Figure 6, Probe 3). It is possible that the genomic form of the X-tail could adopt an open conformation indicating the presence of an unpaired RNA structure [22]. However, we would have expected to see a decrease in signal intensity for Probe 3 following DMS treatment if there was significant unwinding of the X-tail stem-loops.
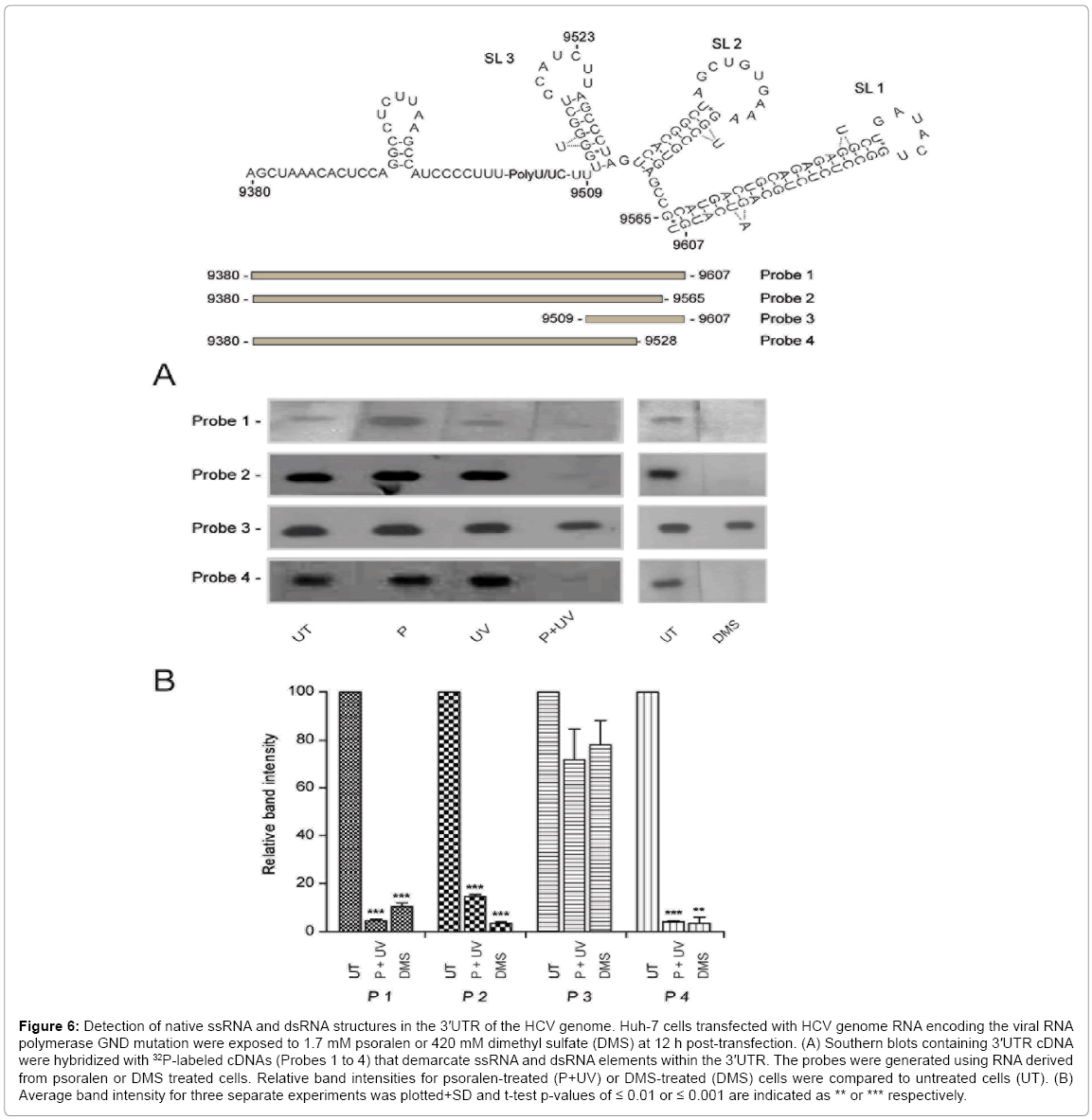
Figure 6: Detection of native ssRNA and dsRNA structures in the 3′UTR of the HCV genome. Huh-7 cells transfected with HCV genome RNA encoding the viral RNA polymerase GND mutation were exposed to 1.7 mM psoralen or 420 mM dimethyl sulfate (DMS) at 12 h post-transfection. (A) Southern blots containing 3′UTR cDNA were hybridized with 32P-labeled cDNAs (Probes 1 to 4) that demarcate ssRNA and dsRNA elements within the 3′UTR. The probes were generated using RNA derived from psoralen or DMS treated cells. Relative band intensities for psoralen-treated (P+UV) or DMS-treated (DMS) cells were compared to untreated cells (UT). (B) Average band intensity for three separate experiments was plotted+SD and t-test p-values of ≤ 0.01 or ≤ 0.001 are indicated as ** or *** respectively.
Since the HCV GND mutant genome used in the in situ psoralencrosslinking experiments was capable of producing a complete set of replication complex proteins, albeit with a non-functional viral RNA polymerase, we tested whether psoralen was blocked from accessing X-tail dsRNA sequences by HCV replication complex proteins. We used a HCV NS2 frame-shift construct that expressed the virion structural proteins including the capsid protein but did not express replication complex proteins NS2, NS3, NS4a/b and NS5a/b. A re-examination of X-tail dsRNA accessibility to psoralen at 12 h post-transfection showed that an absence of viral replication complex proteins restored psoralen crosslinking of X-tail dsRNA (Figure 7, Probe 3).
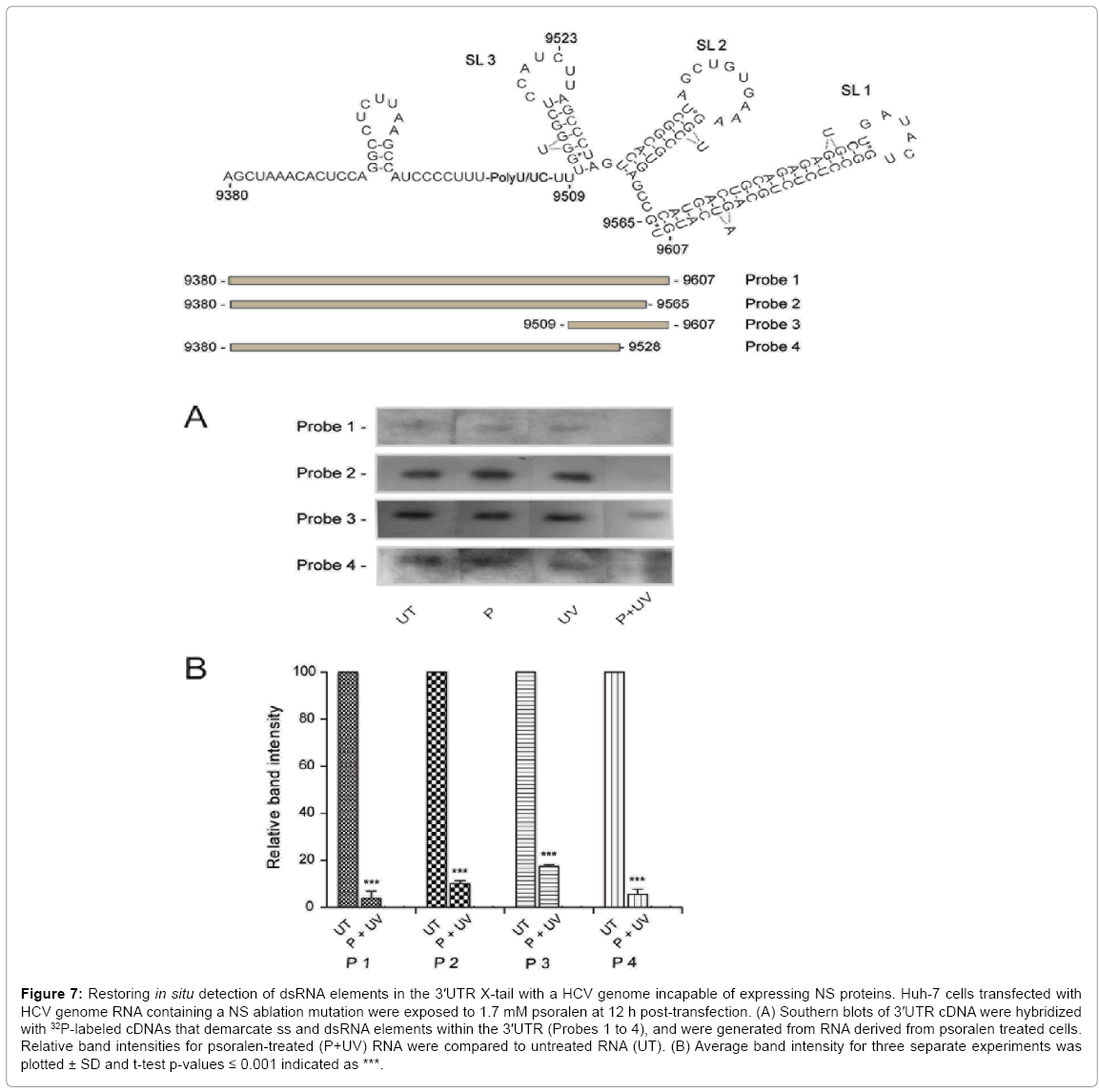
Figure 7: Restoring in situ detection of dsRNA elements in the 3′UTR X-tail with a HCV genome incapable of expressing NS proteins. Huh-7 cells transfected with HCV genome RNA containing a NS ablation mutation were exposed to 1.7 mM psoralen at 12 h post-transfection. (A) Southern blots of 3′UTR cDNA were hybridized with 32P-labeled cDNAs that demarcate ss and dsRNA elements within the 3′UTR (Probes 1 to 4), and were generated from RNA derived from psoralen treated cells. Relative band intensities for psoralen-treated (P+UV) RNA were compared to untreated RNA (UT). (B) Average band intensity for three separate experiments was plotted ± SD and t-test p-values ≤ 0.001 indicated as ***.
An assay system that can preserve authentic intracellular RNA structures to enable differential detection of ssRNA sequences and dsRNA structures in biologically relevant RNAs would be useful for the study of RNA structure-function relationships. We developed a method using in situ chemical crosslinking and nucleotide tagging in combination with site-directed qPCR as a means to detect native ssRNA and dsRNA elements in a low abundance intracellular RNA species. This method was employed here to detect and demarcate ssRNA and dsRNA motifs contained within the 3'UTR sequence of the intracellular HCV genome.
Current structural determination of RNA is highly dependent on whether an in vitro manipulated RNA adopts a form that is biologically relevant or one that is misfolded and trapped into an unproductive meta-stable conformation [13]. In the intracellular environment RNA folding is a dynamic process where initial secondary structures are assembled and disassembled with the aid of protein chaperones before the mature RNA forms its final stable configuration [57]. In the present study we used a full-length and functional 9 kb HCV genome that was refolded in its native setting and complexed with its cognate RNA binding proteins prior to differential tagging with ssRNA and dsRNA modifiers. Current chemical tagging methods, including SHAPE, while of high precision in pinpointing individual unpaired ribonucleotides, generally require large amounts (picomoles) of uniformly-folded starting material prior to chemical tagging and primer extension [12]. This approach would prohibitively preclude the study of low abundance RNAs. The use of in situ nucleotide modification prior to and in concert with RT-PCR and a library of selected primers enabled the detection and demarcation of secondary structures contained within minute quantities of intracellular RNA (Figures 6 and 7). Our mapping precision of ssRNA and dsRNA sequences in the 3'UTR was only limited by the need to construct primers of sufficient sequence specificity and spanning the desired secondary structure under study. Common to all PCR-based technologies, the need to form two sequence-specific annealings generally limits each of the primers in the primer set to 20 or more nucleotides. If a particular secondary structure lies within an RNA sequence that spans the primer set (40 bases), it will not be possible to resolve the nature of the RNA structure.
The HCV 3'UTR is a highly conserved tripartite structure which is essential for the synthesis of the HCV (-) strand RNA intermediate and for virus infectivity [16-18,58]. Attempts to resolve its structure are based largely on subgenomic fragments which have not fully addressed the potential interactions with more distant HCV sequences [53-56]. Our observations for the in vitro modified 3'UTR RNA fragment closely match those obtained for the in situ modified 3'UTR RNA fragment, suggesting that the inherent folding properties of the 3’UTR fragment are not significantly altered by the presence of host cell RNA binding proteins. These results are in line with prior observations and with the predicted model proposing that ssRNA dominates the VR-poly(U/ UC) repeat region both in vitro and in situ, while dsRNA elements are confined to the X-tail (Figure 1) [45].
In order to examine the 3'UTR structure that forms during the initial step of HCV replication, we employed the virus RNA polymerase mutant GND [26]. The HCV GND mutant was expected to maintain HCV RNA in a pre-replicative state and provide a mature and uniform full-length template with which to probe and resolve 3'UTR secondary structures. In contrast to findings for the in vitro and in situ modified 3'UTR fragment, which matched the overall positioning of ssRNA and dsRNA (Figure 5) noted in previous studies [20,22], in situ modification of the HCV genome revealed the presence of a unique dsRNA element in the VR-poly(U/UC) repeat region (Figure 6, Probe 4, P+UV). This suggests that the VR-poly(U/UC) repeat formed a union with an unknown RNA sequence located outside the 3'UTR. Although the VR sequence is not strictly required for in vitro HCV RNA synthesis, its presence imparts a replicative advantage by enhancing 3'UTR-directed (-) strand synthesis [16,59]. An analysis of HCV genotype 1 region for VR-polyU/UC repeat complementary sequences located outside the 3'UTR revealed that the VR segment 9387-ACACUCCAGGCC-9398 located proximal to the small GC stem (Figure 1) could anneal (bold) to a genotype-conserved NS5b sequences 9107-GGCCAGGAGCGU-9119 located to the right of SL9033 [60]. No other sequences in the VR repeat showed significant complementarity to sequences in the HCV genome or to human sequences (http://blast.ncbi.nlm.nih.gov/). While further work is needed to authenticate the formation and function of this VR-NS5b duplex, we speculate that it may form part of a larger RNA replication framework comprised partly of 3'UTR sequences and neighboring NS5b secondary structures [60-63].
The X-tail, which is predicted to contain almost exclusively dsRNA in the form of three dsRNA stem-loops, and the adjacent 26 to 62 polyU/UC repeat sequence constitute the minimal required elements for synthesis of the HCV (-) strand RNA intermediate in vitro [16-18]. The observed occlusion of psoralen by viral replication proteins (Figure 6, Probe 3) and restoration of psoralen access to the X-tail RNA in the absence of viral proteins (Figures 5B and 7, Probe 3) support the view that X-tail stem-loops serve as structural guides which can uniquely capture and align NS proteins during the initial formation of the viral replication complex and prior to synthesis of the viral RNA (-) strand [17].
The capacity to resolve secondary and tertiary RNA structures as present in their native state is key to understanding their individual functions and involvement in intracellular RNA processes. The results presented here serve to elucidate the structural dynamics occurring in the HCV genome during the initial stage of HCV (-) strand RNA replication. The approach used may be potentially extended to the study of native secondary structures in eukaryotic RNAs. The marked differences observed when comparing full-length RNAs with their subgenomic counterparts and the need to provide their native cellular setting reinforces the importance of performing RNA structure analyses in situ, as well as the necessity to use full-size and functional RNAs to form an accurate model of RNA secondary structures. In conclusion, in situ RNA treatment with two complementary RNA modifiers enabling differentiation of native ssRNA and dsRNA structures constitutes a useful tool to improve secondary structure prediction and aid in the identification of tertiary interactions for complex cellular RNA.
This work was funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada # RGPIN 402231. ER was supported through graduate scholarships from the Faculty of Graduate Studies of the University of Montreal.
We thank Dr. Hugo Soudeyns (CHU Sainte-Justine, Canada) for providing the HCV 1b template RNA, and Dr. Tatsuo Takahashi (Health Science Research Resources Bank, Japan) for the Huh-7 human hepatoma cell line.