Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2015) Volume 4, Issue 5
Transthyretin is a homotetrameric thyroid-hormone-transporting protein that binds to the retinol binding protein thus being involved in metabolism, growth, fertility, homeostasis of the cardiovascular and central nervous system, cell differentiation, reproduction, development and maintenance the cognitive processes during aging. Currently, there are several methodologies for natively purifying TTR from plasma, serum, tears, and amyloid fibrils; however, these procedures are laborious. Herein, a low-cost and simple protocol to purify TTR from human plasma is described. It involves the separation of plasma proteins by size exclusion and DEAE chromatography. The homogeneity was assessed by SDS-PAGE and by tandem mass spectrometry using an Orbitrap-XL (Thermo, San Jose-CA).
<Keywords: Transthyretin; Plasma isolation
Transthyretin was discovered in 1942 and originally named as pre albumin [1]; it is a highly conserved protein found in plasma and cerebrospinal fluid [1,2]. It is mainly expressed by hepatocytes [3] and epithelial cells of the choroid plexus [4]; yet it can also be expressed by the cerebral meninges [5], epithelial cells of the retina [6], pancreatic islets of Langerhans [7], visceral yolk sac [8], placenta [9], intestine [10] and at low-scale by stomach, heart muscle, and spleen [11].
The two major physiological functions of TTR are the transport of T4 hormone produced by thyroid and the transport of retinol (vitamin A) through interaction with retinol-binding protein RBP [12], thus being involved in metabolism, growth, fertility, and homeostasis of the cardiovascular and central nervous system [13], cell differentiation, reproduction, development and maintaining the cognitive processes during aging [14].
Previous reports describe evidences of TTR involvement in response to stress [15], in immunological pathways [16], metabolism of lipoproteins [17], and in neuroprotection against Alzheimer's disease by modulating the formation of β-amyloid plaques [18-20]. Paradoxically to this latter function, it is known that when some individuals inherit a mutated TTR gene, the mutated protein loses its functional structure and aggregates. This structural modification leads to formation of amyloid fibers and results in degenerative diseases that affect the nervous system, heart muscle, and other organs. Currently, the molecular mechanisms involved in the conversion of the TTR tetramer into aggregates of amyloid precursor fibers remains elusive. Although it is known that upon dissociation the TTR tetramer produces monomeric species with structural characteristics different from native monomer [21,22], the events associated with this non-native monomer in the initial process of TTR aggregation remain elusive.
The first reports for purifying TTR are from decades ago. Fex andrt Lindgren [23] purified a bovine counterpart to TTR from bovine serum by thiol-disulfide exchange chromatography on thiol- Sepharose 4B and affinity chromatography on human retinol-binding protein linked to Sepharose 4B. Berni et al. [24] purified TTR by using ammonium sulfate fractionation, followed by a hydrophobic interaction chromatography on phenyl-Sepharose and gel filtration on Sephadex G-50. Bashor et al. [25] developed a methodology for TTR purification that involves precipitation of contaminating proteins with dilute aqueous phenol, ion-exchange chromatography on DEAESephacel, and gel permeation chromatography on Sephadex G-100. Furuya et al. [26] were the first to recombinantly produce TTR fused with the E. coli outer membrane protein A (ompA) signal peptide. TTR has also been produced in the eukaryotic Pichia Pastoris system [27]. Lin et al. [28] created a method consisting of serum precipitation, anion exchange, thyroxine affinity chromatography and gel filtration. The latest methodologies for TTR purification include: 1) salting out, anion and cation exchange chromatographies, preparative electrophoresis, and size chromatography [29]; 2) ammonium sulfate fractionation followed by urea/Sephadex G-100 chromatography and a combination of two dye-affinity chromatographic steps on reactive yellow and cibacron blue coupled to agarose columns [30] and 3) extraction of TTR fibrils followed by sequential gel filtration after solubilization in a solution of guanidine hydrochloride and covalent chromatography [31]. This last method aims separating full-length TTR from C-terminal fragments found in TTR amyloid fibrils.
Aiming to establish a simple, fast and efficient method for purification of TTR, this work describes a purification strategy for TTR from blood plasma by employing two chromatographic methods in tandem. In our view, our method is one of the simplest yet for purifying TTR from a natural source; thus, ultimately aiding in studies that focus on the stabilization of the native tetramer for inhibition of their disassembly, prevention of amyloidogenic intermediates formation, inhibition of amyloidogenic intermediates aggregation or promotion of rupture of amyloid fibers [32-34]. A better understanding the TTR properties contribute to the development of more effective therapeutic strategies targeting pathologies related to this protein.
Obtaining human plasma
Blood was collected from volunteer donors in collection tubes containing sodium citrate 3.2%. Blood samples were centrifuged at 15°C, 1500 rpm for 15 minutes. The supernatant (plasma) was collected and the pellet discarded.
Size exclusion chromatography
1 mL of human plasma obtained as described above was used as input for chromatography on Superdex 200 10/300 GL column. Sodium citrate 3.2% pH 8.0 was used as mobile phase for elution. The process was conducted on a FPLC system (Fast Performance Liquid Chromatography-GE-Healthcare). Eluted proteins were boiled in denaturant per 15 minutes and then loaded onto (15% acrylamide) as describe by [35].
Ion exchange chromatography
The fractions from gel filtration containing TTR were pooled and fractionated by ion exchange chromatography on DEAE Sepharose Fast Flow column with Sodium citrate 3.2% pH 8.0 (buffer A) and Sodium citrate 3.2% pH 8.0, 1M NaCl (Buffer B). Elution was performed with a linear gradient of 0-100% B in 10 column volumes with manual hold at each peak. The protein purification was analyzed by SDS PAGE (15% acrylamide) and by tandem mass spectrometry.
LC-MS/MS acquisition
The peptides were subjected to LC-MS/MS analysis using a Thermo Scientific Easy-nLC 1000 ultra-high performance liquid chromatography (UPLC) system coupled online to a LTQ-Orbitrap XL ETD mass spectrometer (Mass Spectrometry Facility-RPT02H PDTIS/ Carlos Chagas Institute-Fiocruz Paraná), as follows. The peptide mixtures were loaded onto a column (75 mm i.d., 15 cm long) packed in house with a 3.2 μm ReproSil-Pur C18-AQ resin (Dr. Maisch) with a flow of 500 nL/min and subsequently eluted with a flow of 250 nL/min from 5% to 40% ACN in 0.5% formic acid and 0.5% DMSO, in a 120 min gradient. The mass spectrometer was set in data-dependent mode to automatically switch between MS and MS/ MS (MS2) acquisition. Survey full scan MS spectra (from m/z 300- 2000) were acquired in the Orbitrap analyzer with resolution R=60,000 at m/z 400 (after accumulation to a target value of 1,000,000 in the linear trap). The ten most intense ions were sequentially isolated and fragmented in the linear ion trap. Previous target ions selected for MS/MS were dynamically excluded for 90 seconds. Total cycle time was approximately three seconds. The general mass spectrometric conditions were: spray voltage, 2.4 kV; no sheath and auxiliary gas flow; ion transfer tube temperature 100°C; collision gas pressure, 1.3 mTorr; normalized energy collision energy using wide-band activation mode; 35% for MS2. Ion selection thresholds were: 250 counts for MS2. An activation q=0.25 and activation time of 30 ms were applied in MS2 acquisitions.
Raw MS data analysis
The reviewed proteome set of Homo sapiens, composed of 20,187 sequences, was downloaded from the UniProt consortium on July 4th, 2014. PatternLab was used for generating a target-decoy database by grouping subset sequences, adding the sequences of 127 common mass spectrometry contaminants, and, for each sequence, including a reversed version of it. The final database used for PSM contained 105,551 sequences.
Peptide sequence matching
The Comet 2014 rev. 1 search engine [36], which is embedded into PatternLab for proteomics [37], was used to compare experimental tandem mass spectra against those theoretically generated from our sequence database and select the most likely peptide sequence candidate for each spectrum. Briefly, the search was limited to fully peptide candidates; we imposed carbamidomethylation of cysteine and oxidation of Methionine as fixed and variable modification, respectively. The search engine accepted peptide candidates within a 40-ppm tolerance from the measured precursor m/z and used the XCorr as the primary search engine score.
PSM validity was assessed using the search engine processor (SEPro) [38], which is embedded in PatternLab version 3.0.0.34. Briefly, identifications were grouped by charge state (+2 and > +3) resulting in two distinct subgroups. For each result, the Comet XCorr, DeltaCN, DeltaPPM, and Peaks Matched values were used to generate a Bayesian discriminator. The identifications were sorted in nondecreasing order according to the discriminator score. A cutoff score was established to accept a false-discovery rate (FDR) of 1% at the peptide level based on the number of labeled decoys. This procedure was independently performed on each data subset, resulting in an FDR that was independent of charge state. Additionally, a minimum sequence length of six amino-acid residues was required. Results were post-processed to only accept peptide spectrum matches with less than 6 ppm from the global identification average and proteins with two or more identified peptides.
Protein relative quantitation by extracted ion chromatograms
Relative quantitation by mass spectrometrydescribes strategies for comparing quantitative information of a same analyte between multiple samples. On the other hand, obtaining absolute quantitation values by mass spectrometry is challenging; some inherent difficulties are: a) different molecules ionize with different efficiencies in the mass spectrometer b) each protein will result in a different number of peptides after tryptic digestion. In a previous report, Zybailov et al described a strategy for normalizing spectral counting quantitation data that provides quantitation values closer to absolute values [39]. We recall that spectral counting is a label-free strategy that correlates the total number of MS/MS spectra assigned to a protein with its relative abundance. Briefly, Zybailov´s normalization, the so-called Normalized Spectral Abundance Factor (NSAF), considers the fact that longer proteins tend to have more peptide identifications that shorter ones. Formally, the NSAF for a protein k is given by “the number of spectral counts (SpC) identifying a protein, k, divided by the protein’s length (L), and divided by the sum of SpC / L for all N proteins in the experiment [39].
In this work, we employed a modified relative quantitation, having roots in NSAF, to assess the effectiveness of our purification approach. Briefly, instead of relying on spectral counts, or strategy, here termed Normalized Ion Abundance Factor (NIAF), replaces spectral count values by extracted ion chromatograms (XIC) values. Briefly, we recall that XICs are obtained by plotting the intensity of a particular peptide ion (m/z) over time and then integrating the area under the curve; for more on quantitative proteomic strategies we refer the reader to [http://dx.doi.org/10.1155/2013/674282]. XICs were obtained by using PatternLab´s SEProQ module [37]. Our motivation in doing so is that protein ratios obtained by XICs yield more accurate estimates ratios than those obtained by spectral counting and therefore yielding estimates closer to the absolute values.
Size exclusion chromatography (SEC), a chromatography that is widely used for purification and determination of the hydrodynamic radius of molecules [40], was the choice as first step for plasma fractionation. Human plasma, when chromatographed through Superdex 200 10/300 column is resolved into five distinct protein peaks (Figure 1).
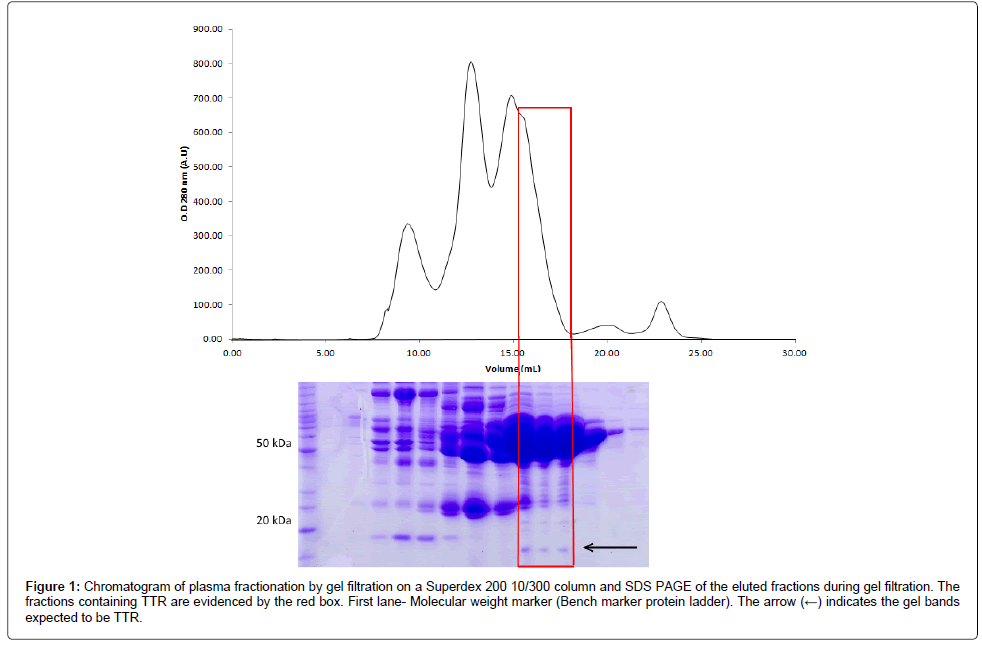
Figure 1: Chromatogram of plasma fractionation by gel filtration on a Superdex 200 10/300 column and SDS PAGE of the eluted fractions during gel filtration. The fractions containing TTR are evidenced by the red box. First lane- Molecular weight marker (Bench marker plasma-Superdex ladder). The arrow (←) indicates the gel bands expected to be TTR.
The SDS-PAGE electrophoresis profile suggested that each peak from superdex 200 10/300 contained different blood proteins. In the fractions corresponding to the shoulder of the third peak, it was possible to verify the presence of a protein with expected size for TTR (Figure 1). Fractions containing the TTR were pooled and submitted to ion exchange chromatography on a DEAE column. IEC resulted in four distinct peaks (Figure 2A). The proteins were well separated and, in the fourth peak, it is possible to indentify TTR by mass spectroscopy with two minor contaminants detected. The active TTR in the Uniprot has ~13.9 kDa and is composed of 127 amino acids. The sequence coverage was 88% and peptides comprising 112 aminoacids out of 127 were detected. We obtained NSIAF from all proteins, as detailed in the methodology; our results showed that 92.5% of the total NSIAF from the sample was decurrently from TTR (Figure 2B). In that same fraction, 4.9% corresponded to the keratin, a common contamination that happens during sample preparation, and 2.4% refers to other proteins.
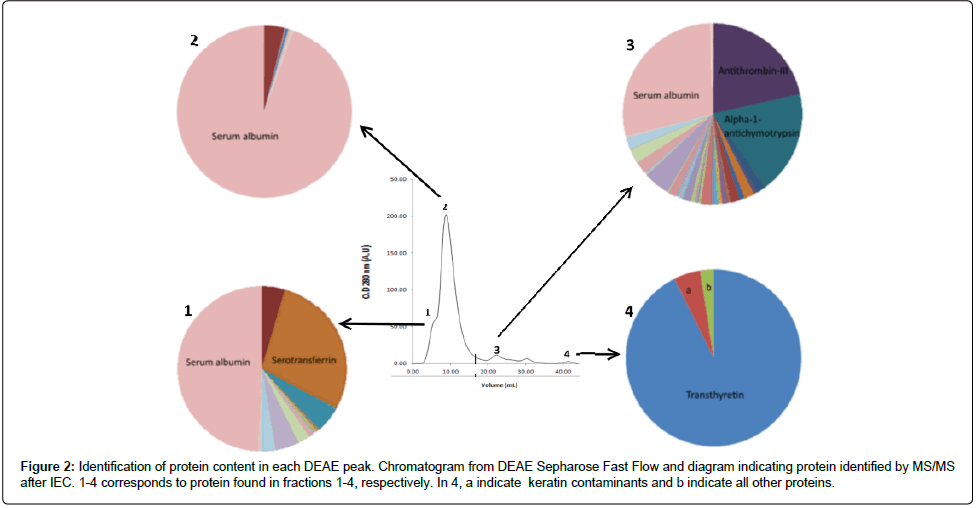
Figure 2: Identification of protein content in each DEAE peak. Chromatogram from DEAE Sepharose Fast Flow and diagram indicating protein identified by MS/MS after IEC. 1-4 corresponds to protein found in fractions 1-4, respectively. In 4, a indicate keratin contaminants and b indicate all other proteins.
All our raw mass spectrometric data is made available at http:// proteomics.fiocruz.br/tatiana/2015/. Moreover, TTR was not detected in any other fraction indicating that all TTR present in plasma is concentrated in this fraction. The yield of purification was ~0.09 mg of protein per ml of plasma. Our experimental procedure also could be helpfull to separate albumin, Serotransferrin and Antithrombin-III from other blood proteins (Figure 2).
Several reports of plasma protein purification by size exclusion chromatography are available; but none were performed with the type of column and buffer system used in this work. By comparing the chromatographic profile of the gel filtration of human plasma presented in Figure 1 with other works, it is possible to observe that the chromatographic profile can vary widely for the same sample at the expense the column used [41,42].
Sepharose 4 FF is widely used for plasma fractionation [43], but due to its low resolution, it is most suitable in cases where the target protein and most contaminants have very distinct molecular masses. Other columns such as Superose 6, Sephadex G-200, Sephadex G-100 also appear in the literature for plasma proteins fractionation [43- 46], but they all have lower resolution than Superdex 200 10/300. Sephadex G-200, the column closest to the required resolution, is capable of providing three distinct peaks when used in the first plasma fractionation step [44]. However, its use at the expense of Superdex 200 10/300, would probably require additional purification steps to obtain pure TTR due to higher amounts of remnant contaminants.
Another important aspect to be noted is that the same column and sample can result in slightly different chromatographic profiles when using buffers with different pH and ionic strength. In this work, the buffer composed of 3.2% Sodium Citrate pH 8.0 was used in all steps of protein purification. The majority of the previously solutions used for plasma purification were buffered with Tris–HCl [23,41,42]. The choice of 3.2% Sodium Citrate was fundamental to better separation of plasma proteins since this buffer prevents the activation of the coagulation cascade, by sequestering calcium, and it is a good buffering at a pH close to physiological, favoring proteins to maintain the conformation found in plasma. Our experimental procedure also could be helpfull to separate albumin, Serotransferrin and Antithrombin-III from other blood proteins (Figure 2). Albumin was found in the second peak of IEC (Figure 2). This protein represents 50% -70% of all plasma proteins [45], and have a remarkable ability to bind to exogenous and endogenous components [46], making its removal important for application related to the remaining plasma proteins.
This method can also effectively be used to isolate two other important proteins: serotransferrin and antithrombin-III (Figure 2). Serotransferrin is associated with Fe3+ uptake, distribution and control of its quantity [47]. Antithrombin-III is an endogenous anticoagulant capable of inhibit key-factors of the coagulation cascade such as thrombin and FXa, thus, having a crucial role in homeostasis [42]. About 25% of the total NSIAF from peak 1 and peak 3 (Figures 2A and 2C) was decurrently from serotransferrin and antithrombin-III, respectively. These proteins can be further purified through additional chromatographic steps. All these results show a simple and low-cost method for chromatographic purification of human plasma TTR. The procedure is also helpfull to remove albumin and to purify two other blood proteins. TTR are often submitted to drug development screenings due to its role in important pathologies such as Senile Systemic Amyloidosis (SSA) and Familial Amyloid Polyneuropathy (FAP) [48] with some promising amyloidogenesis inhibitors [49-54] such as genistein [54,55], other flavonoids of similar structure [55], T4 analogues [49,52,53] being indentified. However all screenings for such compounds were performed with recombinant molecules.