Journal of Antivirals & Antiretrovirals
Open Access
ISSN: 1948-5964
ISSN: 1948-5964
Research Article - (2014) Volume 6, Issue 1
The fundamental concept for developing anti-viral modified nucleoside was proposed. An idea to use 4’-C-substituted-2’-deoxynucleoside derivatives based on the fundamental concept was also proposed to solve the problems of the existing highly active antiretroviral therapy (HAART).
<The highly active anti-retroviral therapy (HAART) has dramatically improved the quality of life and the prognosis of the patients infected by HIV [1,2]. However, the existing HAART has critical problems to be solved. They are (i) emergence of drug-resistant HIV mutants, (ii) drug side effects, and (iii) the need to take large doses of drugs. Therefore, the development of highly potent anti-HIV drugs that prevent the emergence of drug-resistant mutants and have few side effects is required.
The fundamental concept of this study is based on the mutation of viruses. Viruses adapt themselves to the environmental change by mutation. Mutation is that viruses change their genes by taking incorrect (not-programmed) nucleosides into their genes. The fact indicates that the substrate selectivity of viral nucleic acid polymerases is not strict. On the other hand, human beings seldom change their genes. This indicates that the substrate selectivity of human nucleic acid polymerases is very strict. Therefore, by taking the advantage of the difference of the substrate selectivity between viral and human nucleic acid polymerases, it is possible to develop modified nucleosides that are more selectively active to viruses and not active to human beings.
The following working hypotheses were proposed to solve the problems.
The method to prevent the emergence of resistant HIV mutants [Design of 4’-C-substituted-2’-deoxynucleoside (4’SdN) that could Prevent the Emergence of Drug-Resistant HIV Mutants]
All the clinical nucleoside reverse transcriptase inhibitors (NRTIs) belong to the family of 2’,3’-dideoxynucleoside (ddN) (Figure 1).

Figure 1: Structures of Nucleoside Reverse Transcriptase Inhibitor (NRTIs), physiologic 2’-deoxynnucleoside(dN), and 4’-C-substituted-2’-deoxynucleoside (4’SdN).
The ddN structure has been assumed essential for the modified nucleosides to be the chain-terminator of RT. However, resistant HIV mutants against all these drugs emerged very easily and promptly.
The emergence of HIV-mutants resistant to ddNRTIs indicates that the resistant HIV-mutants have obtained the ability to discriminate ddNs from the physiologic 2’-deoxynucleoside (dN) and do not accept the ddNs into the active centre of their RT and/or cut off the incorporated ddNs from the pro-viral DNA terminus.
Therefore, the anti-HIV nucleosides that might prevent the emergence of drug-resistant HIV mutants must satisfy the following two conditions.
1. To prevent the discrimination from dN by HIV, the modified nucleosides should have a structure resembling those of dN as closely as possible so that RT mistakes them for dN.
Since the striking difference of ddN and dN is whether they have 3’-OH, the modified nucleosides must have 3’-OH.
2. In spite of having 3’-OH, the nucleoside must be the chain terminator of RT-catalyzed biosynthesis of pro-viral DNA.
Based on the following hypotheses, 4’SdN (Figure 1) was designed as a nucleoside that could satisfy the above mentioned two conditions.
„„ It would be difficult for HIV to discriminate 4’SdN from dN because 4’SdN has all the functional groups of dN.
„„ The introduction of a substituent at 4’-position makes the 3’- OH into a very unreactive neopentyl-type secondary alcohol. Thus, the 3’-OH of 4’SdN will be used for HIV mistakes 4’SdN for dN, but is too unreactive to be used for the elongation of pro-viral DNA by RT. Therefore, 4’SdN could be the chain terminator of pro-viral DNA biosynthesis.
„„ The steric hindrance between 3’-OH and 4’-substituent changes the conformation of the furanose ring of 4’SdN preferably to the 3’-endo conformation (N-type). This results in 4’SdN being less susceptible to both acidic and enzymatic N-glycolysis than dN and ddN. (In glycolysis, the oxygen atom of the furanose ring participates to form a coplanar oxocarbonium ion, but the conformational change makes it difficult for the oxygen atom to form a coplanar oxocarbonium ion).
„„ Further, the electron-withdrawing 3’-OH makes 4’SdN more acid stable than does ddN even purines. Thus, various purine derivatives can be made in this way.
„„ The lipophilic substituent at 4’-position imparts more lipophylicity to 4’SdNs, thus enabling them to penetrate the cell membrane efficiently. This possibly enhances their bioavailability.
The method to decrease the toxicity of nucleoside
If human DNA polymerase also mistakes 4’SdN for dN, 4’SdN would be highly toxic. However, ddNs, which are the chain terminators of DNA polymerase according to Sanger Method for DNA sequencing [3] and therefore toxic nucleosides, have been used as anti-HIV drugs. These facts mean that RT accepts them as their substrates but DNA polymerase hardly does. Thus, the ability of DNA polymerase to discriminate substrate is superior to that of RT. Therefore, the substrate selectivity between DNA polymerase and RT is different. Thus, by taking the advantage of the difference of the substrate selectivity, it will be possible to develop modified nucleosides which are more selectively active to viruses and less active to human beings than the clinical NRTIs.
The structure of the representative nucleoside antibiotics are shown in Figure 2 [4]. Most of them are nucleoside derivatives modified at one site of the physiologic nucleosides. Though they are highly active against microorganisms, they are highly toxic, too. Therefore, they cannot be clinically used. In the 1960s and 1970s, many organic chemists modified these nucleosides expecting to get nucleoside derivatives having new and/or better biological activity. However, the additional modification of them resulted in the loss or decrease of their activity. The same results were obtained with synthetic modified nucleosides. Namely, highly active one position modified nucleosides are highly toxic, too. The modification of them also resulted in the loss or decrease of their activity. Since the loss and decrease of antibiotic activity means the loss and decrease of toxicity, there is a chance of decreasing the toxicity 4’SdNs by additional modification.

Figure 2: Structures of nucleoside antibiotics.
Examination of the validity of the working hypotheses with 4’-C-methyl nucleosides
On the basis of the working hypotheses, the synthesis and biological evaluation of 4’SdN were carried out. At first, to examine the validity of the working hypothesis, 4’-C-methyl D-ribonucleosides (4’MNs), 4’-C-methyl-2’- deoxynucleosides (4’MdNs), 4’-C-methyl- 2’,3’-dideoxynucleosides (4’MddNs), and 4’-C-methyl-2’,3’- didehydrodideoxynucleosides (4’Md4Ns) (Table 1) were synthesized and evaluated for their biological activity [5,6].
| Structure | Base | EC50(μM) | CC50(μM) | SI(CC50/EC50) |
|---|---|---|---|---|
| 4’MdN | Ad Th Cy Purine |
2.6 7.2 0.072 1.9 |
2.6 104 0.13 >200 |
1.0 - 1.8 >100 |
| 4’Md4N | Ad Th Cy |
>500 21 350 |
>500 330 350 |
~1 16 - |
| 4’MddN | Ad Th Cy |
30 >500 27 |
400 >500 27 |
13 ~1 1 |
| AZT ddA d4T |
0.001 47 4.1 |
>20 >500 >500 |
>2020 >11 >120 |
|
EC50=50% effective concentration; CC50=Cytotoxic concentration; SI-Selectivitiy index (CC50/EC50); 4’MdN=4’=C-methyl-2’-deoxynucleoside; 4’Md4N=4’-C-methyl- 2’-3’-didehydrodi deoxynucleoside; 4’MddN=4’-C-methyl-2’-3’-dideoxy nucleoside; AZT=3’-azido-3’deoxythymidine; ddA=2’,3’-dideoxyadenosine; d4T=2’,3’- didehydro-3’-deoxythymidine
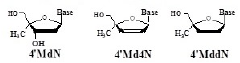
Table 1: Anti-HIV activety of 4’-C-methyl-2’-deoxynucleosides.
4’MdN showed remarkable biological activity (both anti-HIV activity and toxicity), but 4’MddN and 4’Md4N did not show notable biological activity (Table 1).
These results indicate the importance of the 3’-OH for biological activity. Further, we demonstrated that 5’-O-triphosphate of both 4’-C-methyl-2’-deoxycytidine (4’MdC-TP) and 4’-C-methyl-D arabino furanosyl cytidine (4’MAraC-TP) are the chain terminator of calf thymus DNA polymerase α and recombinant rat DNA polymerase β [7]. These results indicate that 4’SdN is NRTI, although further study of 4’MdC-TP with RT was not performed. 4’-C-Methyl-D ribofuranosyl nucleosides (4’MNs) did not show any anti-HIV activity and toxicity at all, because their 5’-OH cannot be phosphorylated by kinase.
Structure-activity relationship (SAR) of 4’SdNs
Next, to study the SAR of 4’SdNs and develop 4’SdNs having more potent anti-HIV activity and less toxicity than 4’MdNs, 4’SdNs having various kinds of 4’-C-substituents and nucleobases were synthesized and evaluated for their biological activity [8-15]. While we were working on our project, the anti-HIV activity of several 4’SdNs was reported by the Syntex group [16-22] and others [23,24]. Therefore, the anti-HIV activities of 4’SdNs that we studied together with those reported by other groups are listed in Table 2.
| Compound | EC50(mM)a) | CC50(mM) | S.I |
|---|---|---|---|
| 4’-C-cyanothymidine | 0.002 | 1 | 500 |
| 4’-C-azidothymidine | 0.01 | 8 | 300 |
| 4’-C-ethynylthymidine | 0.83 | >400 | >482 |
| 4’-C-ethynylarabinofuranosylthymidine | 119 | >400 | >3.4 |
| 4’-C-azidomethylthymidine | 2.1 | 333 | 159 |
| 4’-C-methylthymidine | 7.2 | 104 | 14 |
| 4’-C-ethylthymidine | >400 | 400 | ND |
| 4’-C-methoxythymidine | 8.49 | 200 | 24 |
| 4’-C-vinylthymidine | >400 | >400 | ND |
| 4’-C-hydroxymethylthymidine | 7.0 | >400 | >57 |
| 4’-C-propylthymidine | >100 | >100 | ND |
| 4’-C-cyano-2’-deoxycytidine | 0.0012 | 0.17 | 142 |
| 4’-C-azido-2’-deoxycytidine | 0.004 | 0.21 | 52 |
| 4’-C-ethyny-2’-deoxycytidine | 0.0048 | 2.2 | 458 |
| L-4’-C-ethynyl-2’-deoxycytidine | >400 | >400 | ND |
| 4’-C-ethynyl-2’-deoxy-5-fluorocytidine | 0.030 | >100 | >3333 |
| 4’-C-ethynylarabinofuranosylcytidine | 0.043 | 2.0 | 46.5 |
| 4’-C-methyl-2’-deoxycytidine | 0.015 | 1.0 | 66.7 |
| 4’-C-fluoromethyl-2’-deoxycytidine | 0.0068 | 0.12 | 18 |
| 4’-C-methyl-2’-deoxyadenosine | 2.6 | 2.6 | 1 |
| 4’-C-azido-2’-deoxyadenosine | 0.13 | 50 | 385 |
| 4’-C-ethyny-2’-deoxyadenosine | 0.098 | 16 | 1630 |
| 2’,3’-dideoxy-3’-thia-L-cyrtidine (3TC) | 0.10 | >100 | >1000 |
| 3’-azido-3’-deoxythymidine (AZT) | 0.0032 | 29.4 | 9190 |
a) Anti-HIV activity was determined by MTT assay. MT-4 cells and HIV-1LAI were employed. ND: not determined
Table 2: Anti-HIV activity of 4’-C-substituted-2’-deoxynucleosides.
The SARs of 4’-C-substitued nucleosides against HIV are summarized as follows:
The estimated relative order of anti-HIV activity is as follows:
1. CN ≥ C≡CH > N3 > CH=CH2 > Me= Et > C≡C-CH3. Interestingly, the order is the reverse of the –ΔG° values between equatorial and axial substituents on a cyclohexane ring: CN < F < C≡CH < CH=CH2 < Me ≤ Et < tBu. Thus, these results indicate that the sterically less demanding substituent at the 4’-position gives more potent anti-HIV activity.
2. Purine analogs are generally less toxic than pyrimidine. Although 2’-deoxy-4’-C-ethynyl-5-fluorocytidine, which is a nucleoside derivative modified at two positions of physiologic 2’-deoycytidine, gave a very acceptable Selectivity Index (SI=CC50/EC50) with MT-4 cells, it was toxic with other cells (Kohgo, Yamasa Corporation, private communication ).
3. Arabino analogs are less active and less toxic compared with their corresponding 2’-deoxy counterparts.
4. 4’SddNs do not show high anti-HIV activity.
5. The L-isomers of 4’SdN have no anti-HIV activity,13) although it is known that the L-enantiomer of 2’,3’-dideoxy-3’-thia-Lcytidine (3TC) is as active as the D-enantiomer and less toxic than the D-isomer [24]. This may be due to that the L-isomers are too much modified to be recognized by RT as its substrates.
The biological activity of purine derivatives of 4’-C-Cyano- 2’-deoxy- nucleoside (4’CNdNs) and 4’-C-ethynyl-2’- deoxynucleoside (4’EdNs)
The mentioned results led us to study the biological activity of purine derivatives of 4’CNdN and 4’EdN [25].
The biological activities of them are summarized in Table 3.
| Compound | Base | EC50(μM)a) | CC50(μM) | S.I. |
|---|---|---|---|---|
| 4’-C-Cyano-2’-deoxypurine | A | 0.051 | 12 | 235 |
| I | 0.051 | 23 | 451 | |
| 2AAb) | 0.00079 | 0.034 | 43 | |
| G | 0.000188 | 0.034 | 181 | |
| A | 0.098 | 16 | 1630 | |
| I | 0.15 | 216 | 1440 | |
| 2AA | 0.0003 | 0.82 | 2733 | |
| G | 0.0014 | 1.5 | 975 | |
| AZT | 0.0032 | 29.4 | 9190 | |
a) Anti-HIV activity was determined by MTT assay. MT-4 cells and HIV-1LAI were employed
b) 2-aminoadenine
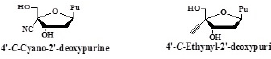
Table 3: Anti-HIV activity of 4’-C-cyano-2’-deoxypurines and 4’-C-ethynyl-2’- deoxypurines.
They are summarized as follows.
1. Some of the purine derivatives of 4’CNdN have high anti-HIV activity, but none of them gives an acceptable SI.
2. All the purine derivatives of 4’EdN have both high anti-HIV activity and acceptable SIs.
Anti-HIV activity of 4’SdNs against drug-resistant HIV mutants [12,13,24]
Many 4’SdNs showed very high anti-HIV activity against wild-type HIV. However, the most important point of our study is whether they are active against drug-resistant HIV-mutants. The anti-HIV activity of selected 4’SdNs against HIV mutants resistant to various NRTIs is listed in Table 4.
| Compound | EC50(μM)a) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HXB2b) | KH65R | L74V | 4l/2l5 | Ml84V | Ml84I | 4l/69/125/SG | MDRd) | Yl8lC | CC50(μM) | |
| 4’EdC | 0.0012 | 0.0008 | 0.0013 | 0.006 | 0.0024 | 0.0026 | 0.015 | 0.0012 | 0.0021 | >200 |
| 4’EaraC | 0.0071 | 0.015 | 0.026 | 0.026 | 0.71 | 0.48 | 0.17 | 0.0079 | 0.016 | >200 |
| 4’MedC | 0.0058 | 0.0071 | 0.0062 | ND | 0.2 | 0.74 | ND | 0.0033 | ND | >200 |
| 4’EdA | 0.008 | 0.0033 | 0.004 | 0.012 | 0.047 | 0.022 | 0.065 | 0.0062 | 0.011 | >200 |
| 4’Ed2AA | 0.0014 | 0.00035 | 0.0007 | 0.0017 | 0.0059 | 0.0027 | 0.0041 | 0.001 | 0.0008 | >200 |
| 4’EdG | 0.007 | 0.001 | 0.0012 | 0.019 | 0.008 | 0.0041 | 0.0068 | 0.0048 | 0.01 | 52 |
| 4’EdI | 0.81 | 0.25 | 0.61 | 1.3 | 1.6 | 1.5 | 2.2 | 0.51 | ND | >200 |
| AZT | 0.022 | 0.02 | 0.02 | 0.3 | 0.01 | 0.017 | 1.6 | 15.3 | 0.014 | >100 |
| 3TC | 0.71 | ND | ND | ND | >100 | >100 | 9.9 | 1.1 | ND | >100 |
| ddC | 0.2 | 3.0 | 1.5 | ND | 2.2 | ND | 1.3 | 5.5 | ND | >100 |
| ddI | 3.9 | 12.7 | 19.5 | 3.6 | 10.1 | ND | 12.2 | 25 | ND | >100 |
Anti-HIV activity was determined with MAGI assay, ND: not determined. b) wild type HIV. d) multidrug-resistant HIV
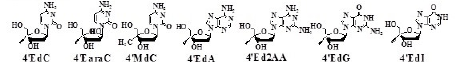
Table 4: Anti-HIV activity of selected 4’SdNs against wild type HIV and drug-resistant HIVs.
It is noteworthy that the three cytidine derivatives maintained their activity against the drug-resistant HIV mutants, although the activity of 4’-C-ethynyl D-arabino-furanosyl cytosine (4’EaraC) and 4’MdC decreased significantly against M184V, M184I, and 41/69/125/ SG. The three purine derivatives, 2’-deoxy-4’-C-ethynyladenosine (4’EdA), 2’-deoxy-4’-C-ethynyl-2- amino adenosine (4’Ed2AA), and 2’-deoxy-4’-C-ethynylguanosine (4’EdG) except for 2’-deoxy-4’-C-ethynylinosine (4’EdI) were highly potent against all drug-resistant HIV-mutants (4’EdI was much less active than the former three derivatives, especially against M184V). Additionally, the three were also active against a non-nucleoside reverse transcriptase inhibitorresistant Y181C. Further, the three purine derivatives were highly potent against the HIVs isolated from seven heavily drug-experienced patients with acquired immune deficiency syndrome (AIDS) as efficiently as against wild-type HIV [14,15,26]. Thus, 4’EdA, 4’Ed2AA, and 4’EdG were highly potent against all the existing HIVs.
These results let us suppose that the three purine 4’EdNs could even prevent the emergence of drug-resistant HIVs. It should be noted that 4’EdG showed toxicity to Hela cells at 52 μM, and therefore, it will be toxic.
Mouse toxicity of purine derivatives of 4’EdNs
Because the three purine derivatives of 4’EdNs showed high activity against all HIVs and acceptable SIs, the mouse toxicity of these 4’EdNs was next examined (Table 5) [25,26].
| Intravenous administration | Oral administration | |||
|---|---|---|---|---|
| Dose (mg/Kg) | Mortality (%) | Dose (mg/Kg) | Mortality (%) | |
| 4’EdA and 4’EdI | 100 10 3 1 |
0 0 0 0 |
100 10 3 1 |
0 0 0 0 |
| 4’Ed2AA | 100 10 3 1 |
100 (1 day)b) 100 (2 days) 0 0 |
100 10 3 1 |
100 (1 day) 100 (2 days) 100 (2 days) 0 |
| 4’EdG | 100 10 3 1 |
100 (1 day) 100 (2 days) 100 (4 days) 0 |
100 10 3 1 |
100 (1 day) 100 (4 days) 100 (4 days) 0 |
a) Six-week-old ICR male mice were employed.
b) Numbers in parentheses represent survival days of mice after administration
Table 5: Toxicity of purine derivatives of 4’-C-ethynyl-2’-deoxynucleosides to micea).
All eight mice survived after a single dosage of 3~100 mgkg-1 of 4’EdA and 4’EdI by both intravenous and oral administrations, but all mice died after a single dosage of 3 mgkg-1 of 4’Ed2AA and 4’EdG irrespective of the administration method (Table 5). Thus, it seemed that 4’EdA and 4’EdI were not toxic, but 4’E2AA and 4’EdG were highly toxic. Thus, 4’EdA seemed very promising.
However, in mice, it was found that 4’EdA and 4’Ed2AA were easily converted to 4’EdI and 4’EdG, respectively, by adenosine deaminase [25,26]. These results showed that the actual toxicity of 4’EdA and 4’Ed2AA to animals is hard to estimate.
Anti-HIV activity of 4’eda derivatives stable to adenosine deaminase
The fact that both 4’EdA and 4’Ed2AA are deaminated by adenosine deaminase prompted us to prepare 4’EdA derivatives stable to the enzyme. It has been known that the adenine derivatives having a halogen atom at the 2-position of the base are stable to adenosine deaminase [27,28]. Therefore, 4’-C-ethynyl-2’-deoxy-2- fluoroadenosine [4’Ed2FA which was later abbreviated as EFdA [29], the structure of EFdA is shown in Table 6, therefore, EFdA is used in this paper], was synthesized and evaluated for the stability to both adenosine deaminase and acidic conditions, and for anti-HIV activity [30,31].
| EFdA | ECldA | Ed4FA | EddFA | Ed4T |
|---|---|---|---|---|
| Compound | Anti HIV activity (Magi assay, μM) | |||
| HIV-1wild | HIV-1MDR | HIV-1M184V | SI | |
| EFdA | 0.00020 | 0.00014 | 0.0031 | 110,000 |
| ECldA | 0.0019 | 0.0084 | 0.01 | 330,000 |
| Ed4FA | 0.80 | 0.15 | 1.8 | |
| EddFA | 0.94 | 8.7 | 97 | |
| AZT | 0.17 | 74.3 | 0.13 | |
| 3TC | 1.0 | 2.8 | >100 | |
| Ed4T | 1.5 | 1.1 | 17 | >50,000 |
| d4T | 7.6 | 64 | 5.6 | |
MAGI = multinuclear activation of galactosesidase indicator, HIV = human immune deficiency virus, AZT = 3’-azido-3’-deoxythymidine; 3TC = 2’,3’-dideoxy-3’-thia-Lcytidime; d4T = 2’,3’-didehydrodideoxythymidine.
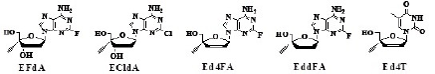
Table 6: Anti HIV activity of 4’-C-substituted-2’-deoxy-2-haloadenosines.
Expectedly, EFdA was very stable to adenosine deaminase under the conditions where 4’EdA was completely deaminated in 60 min (Figure 3) and, further, fairly stable under acidic conditions. Thus, in 120 min only a small part (3%) of EFdA was decomposed under the acidic conditions of gastric juices (pH 1.06) at 24°C, while 2’,3’-dideoxyadenosine (ddA) was completely decomposed in 5 min (Figure 4).

Figure 3: Stability of EFdA to Adenosine Deaminas.

Figure 4: Stability of EFdA at pH 1.06, 36°C.
Because EFdA is a nucleoside derivative modified at two positions (4’-position and 2-position) of physiologic 2’-deoxyadenosine (dA), the toxicity of EFdA is expected to be lower than that of 4’EdA.
While we were working on this project, Haraguchi et.al reported that 4’-C-ethynyl d4T (Ed4T) is more active and less toxic than the clinical d4T and therefore Ed4T is a very promising anti-HIV nucleoside [32]. (The less toxicity is due to additional modification). Therefore, we synthesized dd- and d4-analogs of EFdA and evaluate their anti-HIV activity [33].
The anti-HIV activities of EFdA, 2’,3’-dideoxy-4’-C-ethynyl-2- fluoroadenosine (EddFA) and 2’,3’-didehydrodideoxy-4’-C-ethynyl-2- fluoroadenosine (Ed4FA) together with that of 2’-deoxy-4’-C-ethynyl- 2-chloroadenosine (ECldA) are listed in Table 6 [29].
Although Ed4FA, EddFA, and Ed4T, which do not have 3’-OH, showed some activity against wild-type HIV, they significantly lost any activity against drug-resistant HIVs. EFdA and ECldA showed very high activity against all HIVs and acceptable SIs, however, the activity of ECldA is lower than that of EFdA. These results indicated that the 3’-OH played important roles not only for the phosphorylation of 5’- OH, but also for the activity against drug-resistant HIVs [34].
The most resistant HIV mutant against EFdA emerged for the last 15 years is M184V/T165R/I142, which is 22 times more resistant than wild type HIV [35,36]. Thus, EFdA is sufficiently active against this mutant and has prevented the emergence of resistant mutant for the last 15 years.
Toxicity of EFdA to mice and inhibition of DNA polymerases
Because EFdA is stable to adenosine deaminase and highly active against all HIVs, its mouse toxicity was examined [29,30,33].
EFdA did not show any acute toxicity to mice by either oral or intravenous administration up to 100 mgkg-1 (Figure 5 and Table 7).

Figure 5: Body weight change of mice after a single dosage of 2’-deoxy-4’-Cethynyl-2-fluoroadenosin, administrated orally or intravenously to ICR mice.
| Dose (mgKg-1) | Survivors/total | |
|---|---|---|
| p.o | i.v. | |
| Placebo | 8/8 | 8/8 |
| 1 | 8/8 | 8/8 |
| 3 | 8/8 | 8/8 |
| 10 | 8/8 | 8/8 |
| 30 | 8/8 | 8/8 |
| 100 | 8/8 | 8/8 |
Table 7: Toxicity of 2’-deoxy-4’-C-ethynyl-2-fluoroadenosine (EFdA) after a single dosage to ICR mice.
It is known that the toxicity of NRTIs to animals is caused by their inhibition of mitochondrial DNA polymerase γ. The 50% effective concentration (EC50) of 2’-deoxy-4’-C-ethyny-2-fluoroadenosine- 5-O-triphosphate (EFdA-TP) to inhibit the incorporation of 2’-deoxyadenosine-5-O-triphosphate (dATP) mediated by human mitochondrion DNA polymerase was 10 μM, which was significantly higher than the 0.2 μM of 2’,3’-dideoxyadenosine-5-O-triphosphate (ddA-TP) [30,35]. The EC50 values of EFdA-TP against DNA polymerase α and β were higher than 200 μM. These results indicate that the DNA polymerases scarcely recognize EFdA-TP, a derivative modified at two positions of physiologic dATP, as their substrate but that RT does [30,35,36].
It should be noted that EFdA is highly active to Simian Immunodeficiency Virus (SIV) and did not show any detectable side effects to macaques within 6 months of continuous therapy [37].
Intracellular metabolism of EFdA [35]
The amounts of all fractions of intracellular EFdA metabolites, (EFdA-monophosphate (EFdA-MP), EFdA-diphosphate (EFdADP), and EFdATP) increased proportionately with an increase in the concentration of intracellular EFdA, while compared to AZT-diphosphate and AZT- triphosphate (AZT-TP), only AZT monophosphate markedly increased with an increase in intracellular AZT concentration. The intracellular half-life (T1/2) of EFdA-TP was ~18 h in complete expansion media (CEM) cells, MT4 cells, and multinuclear activation of galactosidase indicator (MAGI)-CCR5 cells (T1/2 of AZT-TP was 3 h). About 50% of the cells were protected against the infection of HIV for 24 h after removal of extracellular EFdA in both MT4 cells and MAGI cells cultured in the presence of 0.1 μM of EFdA.
These results indicate that EFdA, EFdA-DP and EFdA-TP are very stable against intracellular enzymatic catabolism.
A rationalization of the inhibition of RT and DNA polymerase by 4’SdNs
The one position modified 4’SdNs in Figure 6 are highly anti-HIV active and highly toxic, too. These results show that both RT and DNA polymerase accept these one position modified nucleosides. On the other hand, the two positions modified 4’SdNs are highly anti-HIV active but very low toxic. These results show that RT accepts very easily these two positions modified 4’SdNs but DNA polymerase hardly does. These results showed that the substrate selectivity is different between RT (RNA-dependent DNA polymerase) and DNA polymerase (DNA dependent DNA polymerase).

Figure 6: Structures of one position modified 4’SdNs and two positions modified 4’SdNs.
4’-C-ethynyl group has special affinity to RT
The facts that Ed4T is more active than d4T, and that EFdA-TP is two times better substrate for RT than the physiologic substrate 2’-deoxy-ATP [38] had indicated that the 4’-C-ethynyl group will have special affinity to RT. The indication was confirmed first by Yang and his co-workers using Ed4T and X-ray crystallographic method [39]. They showed that the 4’-C-ethynyl group fits into a hydrophobic pocket defined RT residual Ala-114, Try-115, Phe-160, Met-184, and the aliphatic chain of Asp-185.
One year later, the same result was obtained by Michailidis and his co-workers using EFdA [38]. Further, they named EFdA Translocation-Defective Reverse Transcriptase Inhibitor (TDRTI) because the affinity of EFdA to RT by both 4’-C-ethynyl and 3’-OH groups is so strong that the 3’-EFdA-MP-terminated primer strand on the RT does not translocate from the pre-translocation site (N-site) to the post-translocation site (P-site) to accept the next deoxynucleoside triphosphate (dNTP). Therefore, the next dNTP cannot react with the 3’-EFdA-MP-teminus.
Therefore, EFdA has supremely high anti-HIV activity.
The validity of all the working hypotheses is proved and we have developed EFdA, which could prevent the emergence of resistant HIV-mutants, and has the anti-HIV activity of 400 times more active than AZT and several orders of magnitude more active than the other clinical NRTIs, and low toxicity.
Thus, EFdA could solve all the problems of the existing HAART.
Substrate selectivity of viral RNA polymerase is different from that of human RNA polymerase
One of the important findings in our study is that the substrate selectivity of RT (RNA-dependent DNA polymerase) is different from that of human DNA polymerase (DNA-dependent DNA polymerase).
This finding yielded a new question; whether the substrate selectivity of viral RNA polymerase (RNA-dependent RNA polymerase) is different from that of human RNA polymerase (DNA-dependent RNA polymerase).
Eldrup and his co-workers synthesized two position modified 2’-C-methyl-7-deazaadenosine (C), the hybrid of 2’-C-methyl adenosine (A) and the antibiotic tubercidine (B) [40] (Figure 7). The one position modified A is highly active to Hepatitis C Virus (HCV) and highly toxic and the one position modified B is also highly active and highly toxic. They found that the two position modified C is highly anti-HCV active and very low toxic [40]. These results showed that the modification of the highly toxic one position modified nucleosides decreased their toxicity and more significantly that HCV-RNA polymerase accepts the two positions modified but human RNA polymerase hardly does, thus, the substrate selectivity of HCV-RNA polymerase is different from that of human RNA polymerase.

Figure 7: Structures of 4’-C-methyladenosine (A), tubercidine (B), the hybride of A and B [7-deza-2’-C-methyladenoine (C)], 4’-C-azidecytidine (D), 4’-C-azidearaC (E), 4’-C-azido-2’-deoxy-2’-β-fluorocytidine (F), and 4’-C-azide-2’-deoxy-2’,2’-difluorocytidine (G).
On the other hand, Smith and his co-workers reported that 4’-C-azidocytidine (D) is anti-HCV active [41]. This is a striking different point between 4’-C-azidocytidine and 4’-C-alkyl cytidines. (4’-C-alkyl cytidines do not have any biological activity because they cannot be phosphorylated by kinase). Further, they reported that 4’-C-azidoarabinocytidine (E) and 4’-C-azido-2’-deoxy-2’-β- fluorocytidine (F) and 4’-C-azido-2’-deoxy-2’,2’-difluorocytidine (G) (Figure 7) are more active than D and low toxic [41], and further these are chain terminators of HCV-RNA polymerase. Surprisingly, HCV-RNA polymerase accepted these 2’-deoxynucleoside derivatives.
Our preliminary experiments suggested that 4’EdA and its derivative are anti-Flu Virus active (unpublished). These results indicate that the substrate selectivity of viral RNA polymerase is different from that of human RNA polymerase.
The substrate selectivity of viral RNA-dependent nucleic acid polymerases is different from that of human DNA-dependent nucleic acid polymerases. Therefore, by taking the advantage of the difference, it will be possible to develop modified nucleosides which are highly selectively active to viruses and not active to human beings.
The author wishes to express his sincere thanks to the co-workers whose names appear in the cited references for their tremendous efforts to achieve these studies.