Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Research Article - (2023)Volume 13, Issue 6
Objective: In present study, our objective was to discover an mRNA expression signature capable of predicting the Biochemical Recurrence-free (BCR-free) survival of patients with Prostate Cancer (PCa).
Materials and methods: A cohort of 415 patients with pathologically confirmed Prostate Adenocarcinoma (PRAD) from the TCGA dataset was enrolled and analyzed. Using a specific risk score formula, the patients were categorized into high-risk or low-risk groups. Kaplan-Meier survival analyses and Cox regression analyses were conducted to assess the relationship between the mRNA signature and survival outcomes. Furthermore, the KEGG (Kyoto Encyclopedia of Genes and Genomes) analysis was employed to identify potential biological processes and signaling pathways associated with the mRNA signature. To investigate the effects of gene knockdown, CCK8 assay, and transwell assay were employed to explore changes in cell proliferation and invasion ability.
Results and discussion: Eighty-mRNAs showed significant differential expression with a logFC greater than four and a p-value less than 0.05 when comparing biochemical recurrence. Among these, eight mRNAs demonstrated a significant association with Biochemical Recurrence-free (BCR-free) survival. Utilizing a risk score based on the expression levels of these eight mRNAs, we categorized patients into low-risk and high-risk groups, revealing substantial differences in both BCR-free survival and disease-free survival between the two groups. The Oxytocin signaling pathway was involved in this mRNA signature through KEGG analysis. Additionally, cell experiments provided further evidence that the genes within this mRNA signature can influence the proliferation and invasion functions of PCa cells.
Conclusion: In this study, we have successfully developed a novel signature consisting of eight mRNAs, which is valuable in predicting survival outcomes for PCa patients. The clinical implications and underlying mechanisms of these eight mRNAs require further investigation in future studies. These findings open up promising avenues for future research that could lead to a better understanding of the biological significance and therapeutic potential of these mRNAs in PCa patients.
mRNA signature, Prostate cancer, Biochemical recurrence, Survival
Prostate Cancer (PCa) is the most frequently diagnosed cancer and ranks the second leading cause of cancer-related deaths in men in the USA [1]. In China, it is estimated that around 60,300 new cases of PCa and 26,600 PCa-related deaths occurred in 2015 [2]. The standard treatments for PCa include radical prostatectomy, endocrine therapy, radiotherapy, and chemotherapy. Among these, endocrine therapy has been a primary approach in PCa treatment [3]. However, some cases of endocrine therapy-sensitive PCa may progress to Castration-Resistant Prostate Cancer (CRPC), rendering treatment more challenging and leading to a shortened median survival despite the increasing clinical use of new drugs like abiraterone and enzalutamide [4,5].
Due to the complexity and heterogeneity of PCa, accurately predicting the prognosis of individual patients remains a challenge, even with applying parameters such as TNM staging, Gleason score, and PSA values [6]. Men with similar TNM stages, Gleason scores, and PSA values may experience completely different outcomes [7,8]. Therefore, there is a critical need for additional predictive tools and prognostic markers to distinguish between high-risk and low-risk individuals, enabling appropriate clinical management of PCa. Biochemical Recurrence (BCR) monitoring is also crucial in PCa treatment, as it aids in the early detection of recurrence and guides timely interventions [9].
While many studies have demonstrated the association of individual genes, both coding and non-coding RNAs, with biochemical recurrence in prostate cancer, the current research lacks a clear explanation of the underlying mechanisms driving biochemical recurrence [10,11]. Considering that biochemical recurrence is a complex pathological process, relying solely on the expression of a single gene may lead to false-positive results. Recently, identification based on microarray mRNA expression profiles has been developed and extensively used to predict various tumor features and outcomes in different cancer types. However, most studies have focused on individual genes. Our understanding of mRNA characteristics remains limited for prostate cancer, and there are no relevant studies utilizing publicly available datasets to predict biochemical recurrence-free survival. Thus, we conducted this study to delve into the available The Cancer Genomic Atlas (TCGA) dataset to determine if there exists a set of mRNAs that could distinguish between more aggressive phenotypes and poor survival outcomes in PCa patients [12]. We aimed to develop a novel mRNA signature that could significantly enhance the prediction of survival outcomes in prostate cancer patients.
Patients and tissue samples
In this study, we utilized the mRNA expression information from the TCGA Prostate Adenocarcinoma (PRAD) RNA-sequencing database and the complete clinical dataset of TCGA PRAD up to August 11, 2016. These datasets are accessible for download on the UCSC Xena website.
To ensure the reliability and relevance of our analysis, we established specific exclusion criteria. We excluded cases that did not have a histological diagnosis of prostate adenocarcinoma acinar type (n=13 cases) and samples with clinical data but lacking Biochemical Recurrence (BCR) information (n=72 cases). Additionally, we excluded patients with missing essential clinical data, particularly the American Joint Committee on Cancer (AJCC) Tumor Stage (n=7 cases) [13]. Following applying these criteria, we included a total of 415 patients who had both mRNA expression data and corresponding clinical information in our study.
Furthermore, we retrieved mRNA expression data for 59 patients from adjacent non-tumor tissues, serving as the control group for comparative analysis. These datasets form the foundation of our study, allowing us to develop a novel mRNA signature that can effectively predict survival outcomes in prostate cancer patients.
Statistical and data mining analyses of TCGA PRAD mRNA profiles
In this study, we initially screened 27 patients who experienced biochemical relapse within two years and matched them with 140 patients who did not experience biochemical relapse beyond three years using the “matchIt” package. To ensure the two groups' comparability, we used clinic-pathological features, including pathologic T stage, pathologic N stage, pathologic TNM stage, Gleason score [14], residual tumor, primary pattern, and PSA value, with a caliper of 0.05.
After matching, we applied the "limma" package to identify differentially expressed mRNAs based on the criteria of a Fold Change (FC) greater than log2(4) and a p-value less than 0.05. We visualized the differentially expressed mRNAs using a heatmap generated with the "pheatmap" package.
To determine potential mRNAs associated with Biochemical Recurrence-free (BCR-free) survival, we conducted two analyses: logistic regression and single-factor Cox proportional hazards regression—the median expression of the selected mRNAs served as the cut-off point.
Next, we used the following formula to calculate the overall
risk score for the final selected mRNAs: "Risk score= 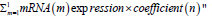 In this formula, "m" represents
the total number of genes in the panel, and "n" is the regression
coefficient obtained from the multivariate Cox regression analysis
for all selected mRNAs. Based on the risk score, we categorized
patients into high-risk or low-risk groups, using the median as the
cut-off point [15].
In this formula, "m" represents
the total number of genes in the panel, and "n" is the regression
coefficient obtained from the multivariate Cox regression analysis
for all selected mRNAs. Based on the risk score, we categorized
patients into high-risk or low-risk groups, using the median as the
cut-off point [15].
Using the log-rank test, we evaluated the survival distribution between these classification groups using Kaplan-Meier survival analysis and assessed the statistical significance between the stratified survival groups. Additionally, we calculated the area under the ROC curve (AUROC) to evaluate the performance of the risk score in predicting BCR-free survival [16].
All data were analyzed using R 4.2.3 packages and SPSS for Windows, version 22. A bilateral p-value of less than 0.05 was considered statistically significant.
GSEA KEGG pathway analysis settings
The utilization of Gene Set Enrichment Analysis (GSEA) involved Java software, and the gene expression file and phenotype file (high/ low-risk group) were prepared following the GSEA guidelines. The parameters were set as 1000 permutations, at least 5 genes in a single pathway, and used the KEGG pathway.
Cell lines and cell culture
The C4-2 cell line, derived from the prostate, was acquired from ATCC (Manassas, VA). These cells were cultured in RPMI 1640 medium (Corning cellgro) supplemented with 10% Fetal Bovine Serum (FBS) from Thermo Fisher Scientific. All cell lines were consistently maintained at 37°C in a 5% CO2 incubator.
Plasmids, antibodies, and reagents: The antibodies utilized in this study were procured from the following companies: Neuropeptide Y, β-Actin (Cell Signaling Technology). The vector of small hairpin RNA (shRNA) is pLKO.1, the sequence of shNPY is selected from sigma, shNPY-#1: GCGACACTACATCAACCTCAT, shNPY-#2: CAGACCTCTTGATGAGAGAAA, shNPY-#3: GTTCCCAGAA CTCGGCTTGAA.
Western blot
Cells were collected and lysed in RIPA buffer (composed of 1xPBS, 1% NP-40, 0.1% sodium dodecyl sulfate, and a protease inhibitor cocktail from Sigma-Aldrich, St. Louis, MO) on ice for more than 15 minutes. The lysate was then centrifuged at 13,000 rpm at 4°C for 10 minutes, and the resulting supernatant was transferred to a new tube for the BCA protein quantification assay using Thermo Fisher Scientific's kit.
Next, 4X loading buffer (from Thermo Fisher Scientific) containing DTT (Dithiothreitol) was added to an equal amount of protein sample and heated at 100°C for 5 minutes. The sample was then subjected to SDS-PAGE analysis and transferred onto a nitrocellulose membrane (Thermo Fisher Scientific).
The membrane was blocked with 5% non-fat milk in 1X TBST (Tris-buffered saline with Tween 20) to prevent nonspecific binding at room temperature for 1 hour. Subsequently, the membrane was incubated with specific primary antibodies at the appropriate dilution at 4°C overnight. Afterward, the membrane was washed three times for 5 minutes each with 1X TBST and incubated with a horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature.
Finally, the immunoblots were visualized using SuperSignal West Pico Stable Peroxide Solution (Thermo Fisher Scientific).
Real-time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from the cells using Trizol reagent (Thermo Fisher Scientific). From 1 μg of total RNA, the first strand cDNA was synthesized using a GoScript kit (Promega). Subsequently, a two-step real-time Polymerase Chain Reaction (PCR) was carried out using SYBR Green Mix (Bio-Rad) and the C1000 Touch Thermal Cycler, CFX96 Real-Time System (Bio-Rad), following the manufacturer's protocol.
All signals were normalized against GAPDH to ensure accurate quantification, and the 2-ΔCt method was applied to calculate the fold change. For the NPY gene, the following primer sequence was used for PCR amplification: Forward: CTGCGACACTACATCAATCTCATCA, reverse: CAGTGTCT CAGGGCTGGATCTC.
Cell viability and migration assays: Cell viability was assessed using the Cell Counting Kit-8 (CK04, DOJINDO, Kumamoto, Japan). For transwell assays, 4 × 104 to 8 × 104 cells were seeded into the upper chamber (CLS3464, Corning Costar, Corning, NY, USA) without FBS supplementation, while the lower chamber was filled with 500 μL DMEM containing 10% FBS. After 36 to 72 hours of culture, migrated cells were fixed using 4% paraformaldehyde (G1101, Servicebio, Wuhan, Hubei, China), stained with Crystal Violet Staining Solution (C0121, Beyotime, Shanghai, China), and subsequently counted under a microscope.
Patient characteristics
Based on pathology, all 415 patients included in our study were confirmed to have Prostate Cancer (PCa). The mean age at diagnosis was 60.88 years (standard deviation, SD=6.952), and the average follow-up period was 20.713 months (Table 1). Notably, 52 patients with adjacent non-tumor tissue were not part of our screening for differential expression of mRNAs between PCa and non-tumor tissue. Throughout the follow-up period, 36 patients experienced biochemical recurrence.
| Characteristics | TCGA cohort (N = 415) | |
|---|---|---|
| N | % | |
| Age, median (range) | 61 | 41–78 |
| Biochemical recurrence | ||
| Yes | 36 | 8.7 |
| No | 379 | 91.3 |
| pT stage | ||
| T2 | ||
| T2a | 11 | 2.7 |
| T2b | 10 | 2.4 |
| T2c | 152 | 36.6 |
| T3 | ||
| T3a | 131 | 31.6 |
| T3b | 103 | 24.8 |
| T4 | 8 | 1.9 |
| pN stage | ||
| N0 | 293 | 70.6 |
| N1 | 61 | 14.7 |
| Nx | 61 | 14.7 |
| Gleason score | ||
| 6 | 42 | 10.1 |
| 7 | 214 | 51.6 |
| 8 | 52 | 12.5 |
| 9 | 104 | 25.1 |
| 10 | 3 | 0.7 |
| Pathologic stage | ||
| II | 178 | 42.9 |
| III | 172 | 41.4 |
| IV | 65 | 15.7 |
| Residual tumor | ||
| R0 | 266 | 64.1 |
| R1 | 119 | 28.7 |
| R2 | 5 | 1.2 |
| Rx | 25 | 5.1 |
| Radiation therapy | ||
| Yes | 52 | 12.5 |
| No | 363 | 87.5 |
Table 1: Clinicopathological characteristics of the patients with prostate cancer.
Differentially expressed mRNA of PCa between patients with/without biochemical relapse
This study analyzed mRNA expression in two groups: 10 prostate cancer tissues with biochemical relapse and 10 without biochemical relapse. Eighty differentially expressed mRNAs were identified, with a fold change greater than log2(4) and a p-value less than 0.05. Among these 83 mRNAs, 18 (21.7%) were found to be downregulated, while the remaining 65 (78.3%) were upregulated, as summarized in Table 2.
| Sample ID | logFC | AveExpr | t | P.Value | adj.P.Val | B |
|---|---|---|---|---|---|---|
| CYP3A5 | 2.48239 | 4.569375 | 5.652625 | 1.97E-05 | 0.154648 | 2.404649 |
| SNAP91 | 3.20046 | 6.2164 | 5.546416 | 2.48E-05 | 0.154648 | 2.229532 |
| KNDC1 | 2.21337 | 4.242665 | 4.907949 | 0.000101 | 0.257873 | 1.139164 |
| D4S234E | 2.49082 | 6.24882 | 4.689008 | 0.000164 | 0.257873 | 0.751892 |
| STXBP5L | 2.41183 | 4.665825 | 4.631881 | 0.000187 | 0.257873 | 0.649876 |
| DAPL1 | 3.60477 | 2.317155 | 4.476141 | 0.000265 | 0.257873 | 0.369917 |
| PROK1 | 2.5193 | 5.14597 | 4.462068 | 0.000274 | 0.257873 | 0.344495 |
| CCKBR | 2.20471 | 2.114085 | 4.411507 | 0.000307 | 0.257873 | 0.253 |
| RPE65 | 2.8301 | 2.27617 | 4.120485 | 0.000594 | 0.257873 | -0.27784 |
| FOXA2 | 2.73762 | 1.94139 | 4.045647 | 0.000704 | 0.257873 | -0.41529 |
| SMOC1 | 2.49096 | 7.92976 | 4.04544 | 0.000704 | 0.257873 | -0.41567 |
| DACT2 | 3.50009 | 6.851395 | 3.996573 | 0.000787 | 0.257873 | -0.50557 |
| CPNE6 | 3.28618 | 3.29978 | 3.992231 | 0.000794 | 0.257873 | -0.51357 |
| COL27A1 | 2.19281 | 6.508175 | 3.955879 | 0.000863 | 0.257873 | -0.58052 |
| JPH4 | 2.33513 | 7.086825 | 3.954687 | 0.000865 | 0.257873 | -0.58272 |
| S1PR5 | 2.17338 | 4.15609 | 3.795804 | 0.001241 | 0.266903 | -0.87585 |
| DMKN | 2.23259 | 6.194565 | 3.71275 | 0.001499 | 0.271769 | -1.02926 |
| CA14 | 2.60907 | 3.475315 | 3.673135 | 0.00164 | 0.271769 | -1.10244 |
| P2RX1 | 2.05353 | 6.005505 | 3.669818 | 0.001652 | 0.271769 | -1.10856 |
| CYP4F8 | 2.89756 | 4.62811 | 3.622443 | 0.00184 | 0.274265 | -1.19606 |
| VWDE | 2.21256 | 3.91715 | 3.475872 | 0.002563 | 0.280561 | -1.46644 |
| CDH22 | 2.05477 | 3.520055 | 3.457826 | 0.00267 | 0.280561 | -1.49968 |
| SP5 | -2.10771 | 7.457765 | -3.44124 | 0.002771 | 0.280561 | -1.5302 |
| BCL11A | 2.07122 | 5.28653 | 3.349779 | 0.003405 | 0.280561 | -1.69832 |
| NLRP8 | -2.22509 | 3.299375 | -3.25975 | 0.004168 | 0.284259 | -1.86324 |
| CYP4F22 | 2.10217 | 2.948215 | 3.253942 | 0.004222 | 0.285428 | -1.87385 |
| C20orf56 | 2.98507 | 2.868715 | 3.253244 | 0.004229 | 0.285428 | -1.87512 |
| KIAA1210 | 2.36534 | 6.51733 | 3.202225 | 0.00474 | 0.290774 | -1.96824 |
| LAMA1 | -2.21471 | 4.836365 | -3.19839 | 0.004781 | 0.291228 | -1.97522 |
| SEMG2 | 3.2885 | 2.78095 | 3.19664 | 0.004799 | 0.29123 | -1.97842 |
| HRNBP3 | 2.65739 | 6.479695 | 3.157145 | 0.005241 | 0.304962 | -2.0503 |
| IP6K3 | 2.27714 | 4.26498 | 3.128646 | 0.005584 | 0.313589 | -2.10206 |
| LRAT | 2.15433 | 3.860085 | 3.059019 | 0.006518 | 0.332346 | -2.2281 |
| UNC5A | -3.74871 | 6.844265 | -3.01636 | 0.007163 | 0.338903 | -2.30502 |
| TGM3 | -2.23767 | 7.246545 | -2.99912 | 0.00744 | 0.343076 | -2.33601 |
| NTF4 | 2.07247 | 3.612355 | 2.994134 | 0.007523 | 0.343275 | -2.34498 |
| KCNJ3 | 2.35939 | 3.847745 | 2.988817 | 0.007611 | 0.343275 | -2.35453 |
| OGDHL | 2.3637 | 5.97416 | 2.975557 | 0.007837 | 0.343275 | -2.37832 |
| WFDC2 | 2.26027 | 7.957015 | 2.97496 | 0.007847 | 0.343275 | -2.37939 |
| HNF1B | 2.33896 | 6.18425 | 2.951309 | 0.008267 | 0.347773 | -2.42177 |
| NTNG1 | 2.48337 | 6.310835 | 2.930293 | 0.008657 | 0.353654 | -2.45935 |
| EPHA5 | 2.42475 | 4.840035 | 2.909116 | 0.009069 | 0.353768 | -2.49714 |
| NRCAM | -2.11698 | 7.44729 | -2.89629 | 0.009327 | 0.353768 | -2.52 |
| CAPN9 | -2.40501 | 4.561695 | -2.80736 | 0.011323 | 0.369721 | -2.6776 |
| C1orf64 | -3.98135 | 6.997025 | -2.79735 | 0.011572 | 0.371318 | -2.69524 |
| DIO1 | -2.88787 | 4.193225 | -2.78123 | 0.011984 | 0.373552 | -2.72362 |
| TRIM29 | 2.24354 | 8.5821 | 2.776437 | 0.012109 | 0.373552 | -2.73205 |
| NKAIN1 | 2.81266 | 7.36192 | 2.74063 | 0.013084 | 0.379372 | -2.79484 |
| CEACAM20 | 3.33379 | 5.432085 | 2.7329 | 0.013304 | 0.38097 | -2.80836 |
| DLK2 | 2.04829 | 5.329305 | 2.699952 | 0.014283 | 0.386302 | -2.86584 |
| LAMB3 | 2.11984 | 8.25956 | 2.695571 | 0.014418 | 0.386369 | -2.87346 |
| WISP2 | 2.02771 | 5.631785 | 2.675632 | 0.015049 | 0.386694 | -2.9081 |
| LIX1 | 2.26533 | 3.387965 | 2.660976 | 0.015529 | 0.390952 | -2.93351 |
| SEMG1 | 2.95404 | 2.15583 | 2.637217 | 0.016339 | 0.396964 | -2.97458 |
| STAC2 | 2.07672 | 4.10782 | 2.629181 | 0.016621 | 0.396964 | -2.98844 |
| CST4 | -2.65467 | 3.657695 | -2.59482 | 0.017884 | 0.403036 | -3.04751 |
| GSTA3 | -2.35385 | 1.637865 | -2.58152 | 0.018396 | 0.403036 | -3.07029 |
| LOC572558 | 2.22935 | 5.288055 | 2.579946 | 0.018458 | 0.403036 | -3.07299 |
| LOC100190940 | 2.709 | 3.36981 | 2.575164 | 0.018646 | 0.404655 | -3.08117 |
| PSCA | -3.09858 | 11.32337 | -2.55876 | 0.019306 | 0.404891 | -3.10918 |
| ZIC2 | -2.50334 | 5.21855 | -2.55035 | 0.019652 | 0.407005 | -3.12351 |
| SP8 | 2.0091 | 6.03122 | 2.54842 | 0.019733 | 0.407005 | -3.1268 |
| SLC6A14 | -2.09713 | 6.402185 | -2.52153 | 0.020885 | 0.408366 | -3.17247 |
| CYP26A1 | -2.14378 | 1.87719 | -2.51919 | 0.020989 | 0.408366 | -3.17644 |
| NPY | 4.36698 | 10.05492 | 2.5036 | 0.021689 | 0.408366 | -3.20283 |
| LRRC7 | 2.11347 | 4.557115 | 2.464883 | 0.023524 | 0.420709 | -3.26803 |
| POTEH | 2.00032 | 3.96211 | 2.411235 | 0.026308 | 0.434875 | -3.35764 |
| TBX10 | -2.46335 | 3.564355 | -2.40907 | 0.026427 | 0.435784 | -3.36125 |
| TRPM8 | 2.37912 | 12.6442 | 2.397397 | 0.027075 | 0.43764 | -3.38061 |
| SFTPA2 | -2.53079 | 10.53594 | -2.37769 | 0.028203 | 0.442484 | -3.41321 |
| TP63 | 2.15541 | 7.124175 | 2.374184 | 0.028408 | 0.442484 | -3.419 |
| SCUBE2 | 2.22925 | 8.863045 | 2.35789 | 0.02938 | 0.445468 | -3.44584 |
| FLJ26850 | 2.0284 | 4.4705 | 2.354599 | 0.02958 | 0.446456 | -3.45125 |
| CLSTN2 | 2.35366 | 5.86712 | 2.317728 | 0.031908 | 0.459234 | -3.51161 |
| LOC642587 | 2.2652 | 6.14139 | 2.304628 | 0.032776 | 0.461156 | -3.53295 |
| ACTC1 | 2.52549 | 7.316945 | 2.286287 | 0.034027 | 0.465099 | -3.56272 |
| KRT14 | 2.27605 | 7.428815 | 2.280263 | 0.034448 | 0.46585 | -3.57247 |
| CEACAM22P | 2.25249 | 3.381415 | 2.278702 | 0.034557 | 0.465966 | -3.575 |
| CARTPT | 2.16944 | 3.2462 | 2.262136 | 0.035742 | 0.472952 | -3.60174 |
| EEF1A2 | -2.06218 | 10.10811 | -2.24718 | 0.036844 | 0.479038 | -3.62579 |
| CLEC3A | 2.28827 | 2.307475 | 2.215947 | 0.039246 | 0.487719 | -3.67576 |
| KRT13 | 2.65958 | 5.17372 | 2.187931 | 0.041521 | 0.500843 | -3.72027 |
| NEFL | 2.70225 | 4.281065 | 2.126824 | 0.046909 | 0.516417 | -3.81627 |
Table 2: The mRNA after limma package filter for FC>4 and P<0.05.
Establishment of mRNA signatures associated with survival in PCa patients
Our study aimed to identify potential mRNA markers with prognostic significance for Biochemical Recurrence-free survival (BCR-free) in prostate cancer patients. We conducted rigorous analyses using binomial logistic regression and univariate Cox proportional hazards regression on the differentially expressed mRNAs to achieve this. From these analyses, we identified eight mRNAs that demonstrated a statistically significant association with BCR-free survival at a significance level of<0.05. Among these mRNAs, SCUBE2 exhibited a positive correlation with BCR-free survival, while the other seven mRNAs (IP6K3, CA14, LIX1, EPHA5, HRNBP3, RPE65, and NPY) displayed a negative correlation, as presented in Figure 1.

Figure 1: Kaplan–Meier estimates of RFS of patients with PRAD in eight genes signature.
By establishing the median risk score as the critical value, we successfully stratified the patients into a high-risk group of 208 patients and a low-risk group comprising 207 patients. Importantly, we observed statistically significant differences between the two groups in key clinicopathological features, such as T stage, lymph node status, pathological stage, Gleason score, and marginal distribution (Table 3). However, age at diagnosis did not significantly differ between the groups.
| Variable | Low-risk n (%) 207 |
High-risk n (%) 208 |
Pearson X2 | P value |
|---|---|---|---|---|
| Age of diagnosis | 0.525 | 0.469 | ||
| ≤ 65 | 150 | 144 | ||
| >65 | 57 | 64 | ||
| AJCC T stage | 30.442 | < 0.001 | ||
| T2 | 144 | 59 | ||
| T3 + T4 | 93 | 149 | ||
| Lymphonodus status | 20.743 | < 0.001 | ||
| Negative | 193 | 161 | ||
| Positive | 14 | 47 | ||
| Pathological stage | 37.552 | < 0.001 | ||
| Stage II | 116 | 62 | ||
| StageIII | 76 | 96 | ||
| StageIV | 15 | 50 | ||
| Gleason Score | 42.57 | < 0.001 | ||
| <8 | 160 | 96 | ||
| ≥8 | 47 | 112 | ||
| Residual Tumor | 4.148 | 0.042 | ||
| R0 | 141 | 125 | ||
| others | 52 | 72 | ||
| Radiation therapy | 14.6665 | < 0.001 | ||
| Yes | 184 | 160 | ||
| No | 13 | 39 |
Note: * Statistical significant results.
Table 3: Correlations between risk score of eight-mRNA signature and clinicopathological characteristics.
Additionally, we assessed the validity of the 8-mRNA signature in predicting Relapse-Free Survival (RFS) by comparing the lowrisk and high-risk score groups. Our analysis demonstrated a clear and significant difference between the two groups (P=0.001). Furthermore, even in a multivariate Cox regression analysis, the 8-mRNA signature remained an independent prognostic factor for RFS, regardless of the Gleason score (Figure 2 and Table 4).

Figure 2: Analysis of 8-mRNA risk score in TCGA patients. The distribution of 8-mRNA risk score, RFS status and mRNA expression profiles were analyzed in TCGA patients. Note: A) 8-mRNA genes risk score distribution; B) Patient’s status and time; ( ) No recurrence; (
) No recurrence; ( ) Recurrence . The dashed line in the middle of a, b divided the patients into low-risk and high-risk groups; C) Heat map of six autophagy-related genes expression profiles; D) Kaplan-Meier estimation of overall survival in prostate cancer patients using 8-mRNA genes in the TCGA signature dataset; ( ) Low Risk; ( ) High Risk.
) Recurrence . The dashed line in the middle of a, b divided the patients into low-risk and high-risk groups; C) Heat map of six autophagy-related genes expression profiles; D) Kaplan-Meier estimation of overall survival in prostate cancer patients using 8-mRNA genes in the TCGA signature dataset; ( ) Low Risk; ( ) High Risk.
| Variable | Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P value | HR | 95% CI | P value | |
| Risk score (high vs. low) | 2.722 | 1.945–3.809 | <0.001 | 2.024 | 1.398–2.930 | <0.001 |
| Age | 1.009 | 1.007–1.046 | 0.68 | 0.992 | 0.950–1.305 | 0.697 |
| T stage | ||||||
| T2 | As a comparison | As a comparison | 0.011 | As a comparison | As a comparison | 0.814 |
| T3 | 2.216 | 1.358–5.040 | 0.004 | 0.804 | 0.143–4.519 | 0.805 |
| T4 | 3.824 | 0.849–17.224 | 0.081 | 1.301 | 0.150–11.290 | 0.811 |
| N stage | 1.777 | 0.915–3.452 | 0.089 | |||
| Pathological stage | ||||||
| II | As a comparison | As a comparison | 0.002 | As a comparison | As a comparison | 0.416 |
| III | 3.609 | 1.647–7.909 | <0.001 | 2.311 | 0.360-14.855 | 0.377 |
| IV | 4.452 | 1.826–10.856 | <0.001 | 1.461 | 0.245–8.699 | 0.677 |
| Gleason Score (< 8 vs. ≥8) | 5.428 | 2.835–10.396 | <0.001 | 2.853 | 1.354–6.010 | <0.001 |
| Residual Tumor (R0 vs. others) | 2.688 | 1.524–4.472 | <0.001 | |||
| Radiation Therapy | 2.252 | 1.146–4.424 | 0.019 | |||
Note: HR-hazard ratio; CI-Confidential interval; vs.- versus; *Statistical significant results.
Table 4: Univariate and multivariate Cox regression analyses of eight-mRNA signature in the prediction of RFS.
Although the 8-mRNA-based risk score did not show a significant association with AJCC T stage in the multivariate analysis, when we stratified patients based on T stage (T0-2 vs. T3-4; Figure 3a, P<0.001, unpaired t-test), we found a positive correlation with this signature. Similarly, we observed higher risk scores in the group with high Gleason scores (GS<8 vs. GS ≥ 8; Figure 3A, P<0.001, unpaired t-test). Thus, we conducted a subgroup analysis to investigate the predictive value of the 8-mRNA signature across different AJCC T stages and Gleason scores (Figure 3B).

Figure 3: (A) Boxplot of risk score in patients with different T stage. ( ) 2; ( ) >2; (B) Boxplot of risk score in patients with different Gleason Score. ( )≤7; ( )≥8.
Finally, we employed the ROC package to compare the predictive accuracy of the multivariate logistic model with the 8-mRNA signature and TNM stage. Our results revealed that incorporating the 8-mRNA signature into the model led to a substantial improvement in accuracy for predicting 2-year and 5-year Overall Survival (OS) by 15.7% and 11.5%, respectively (AUROC 0.715 vs. 0.558, 0.747 vs. 0.632, Figure 4). These findings underscore the potential of the 8-mRNA signature as a valuable tool for predicting survival outcomes in prostate cancer patients.

Figure 4: ROC curves of the multivariate logistic regression model with risk score of the six autophagy-related genes signature or with pathologic stage in prediction of 2-year and 5-year RFS.
mRNA signature-associated signaling pathways
This study utilized Gene Set Enrichment Analysis (GSEA) using the Java software from the Broad Institute to identify relevant signaling pathways and biological processes in PRAD. Figure 5 demonstrates the enrichment of specific cancer-associated pathways, including the cell cycle, NOD-like receptor signaling pathway, and Toll-like receptor signaling pathway, in the high-risk patient group. These findings suggest a potential involvement of the 8-mRNA signature in tumor growth and immune escape in PRAD.

Figure 5: Gene Set Enrichment Analysis (GSEA) of the 8-mRNA genes signature in TCGA dataset. Note: A) The significant 10 biological processes and signaling pathway; (B) GSEA validated enhanced activity of cell cycle.
NPY inhibited PCa cell growth and metastasis
Based on the KEGG pathway analysis mentioned earlier, it was established that the signature is implicated in various functions, including the cell cycle. This led us to hypothesize that the genes within this signature may significantly impact the proliferation, invasion, and metastasis of Prostate Cancer (PCa) cells. As a result, we decided to focus on the NPY gene from the signature for further investigation.
To explore the functional role of NPY, we conducted experiments involving the knockdown of NPY expression in C4-2 cells (as depicted in Figures 6A and 6B). Subsequently, transwell experiments were performed, and the results provided evidence that NPY knockdown enhanced the invasive ability of C4-2 cells (Figures 6C and 6D). Moreover, we observed that NPY knockdown also augmented the proliferation capacity of C4-2 cells (Figure 6E).

Figure 6: Changes in cell proliferation and invasion function after knockdown of NPY gene in C4-2 cells. Note: (A) The verification of knockdown efficiency was performed in western blot; (B) qPCR respectively. ( ) sh-NC; (
) sh-NC; (  ) sh-NPY-#1; (
) sh-NPY-#1; (  ) sh-NPY-#2; (
) sh-NPY-#2; (  ) sh-NPY-#3; C and D) The invasive ability of C4-2 cells knocked down by NPY was enhanced. (
) sh-NPY-#3; C and D) The invasive ability of C4-2 cells knocked down by NPY was enhanced. ( ) sh-NC; (
) sh-NC; ( ) sh-NPY-#1; (
) sh-NPY-#1; ( ) sh-NPY-#2; ( ) sh-NPY-#3 ; E) The proliferation ability of C4-2 cells knocked down NPY was enhanced. (
) sh-NPY-#2; ( ) sh-NPY-#3 ; E) The proliferation ability of C4-2 cells knocked down NPY was enhanced. (  ) sh-NC; (
) sh-NC; (  ) sh-NPY-#1; (
) sh-NPY-#1; ( ) sh-NPY-#2; (
) sh-NPY-#2; ( ) sh-NPY-#3.
) sh-NPY-#3.
These experimental findings support the notion that NPY plays a significant role in regulating the invasive and proliferative functions of C4-2 cells in prostate cancer. The results highlight the potential importance of NPY in the context of prostate cancer progression and suggest its possible relevance as a therapeutic target for managing the disease.
Considering the heterogeneous nature of Prostate Cancer (PCa), achieving accurate risk assessment and effective management of patients after Radical Prostatectomy (RP) poses a significant challenge [17]. One crucial clinical issue PCa researchers face is identifying and developing reliable biomarkers that can aid in determining whether prompt adjuvant therapy is necessary following surgical or radiotherapeutic intervention [18].
The present study focused on the clinical relevance of mRNAs in prostate cancer. It investigated their involvement in complex biological functions observed in various cancer types, including Prostate Adenocarcinoma (PRAD), such as cell cycle regulation and immune responses. Specifically, we identified several mRNAs, including IP6K3 [19,20], HRNBP3 [21], CA14 [22-24], LIX1 [25,26], RPE65 [27,28], EPHA5 [29], NPY [30-33], and SCUBE2 [34-36], which were associated with higher Gleason scores, advanced TNM staging, disease progression, metastasis, or unfavorable survival outcomes. By combining these candidate mRNAs, we improved our results' accuracy and reduced the impact of confounding factors. Recent advancements have led to the development of RNA sequencing-based signatures in various cancers, such as liver cancer, to identify subgroups exhibiting aggressive phenotypes or poor survival outcomes. Applying such signature-based approaches can enhance risk stratification and treatment decision-making in prostate cancer and other malignancies [37].
Despite the progress made in constructing mRNA signatures for cancer prognosis, many of these signatures have shown limitations in accurately reflecting clinical features and aligning with the specific needs of patients. Our study detected an mRNA signature by comparing tumor tissue with adjacent tissues in the study participants. However, this signature is needed to capture the observed clinical features fully.
To the best of our knowledge, our study is the first to reveal cases where non-Biochemical Recurrence (non-BCR) resulted in recurrence or metastasis. At the same time, patients with BCR experienced a more favorable prognosis. Within these two groups, we identified a set of 83 differentially expressed mRNAs. By analyzing RNA sequencing data from 415 prostate cancer patients, we identified an 8-mRNA signature that correlated with the AJCC T stage and predicted worse patient outcomes.
Moreover, through further multivariate analysis, we demonstrated that this 8-mRNA signature was an independent predictor of Recurrence-Free Survival (RFS) in patients with prostate cancer. These findings highlight the potential clinical significance of this signature in risk stratification and prognosis assessment, providing valuable insights into its utility as a predictive tool in the management of prostate cancer patients.
To assess the independent predictive value of the 8-mRNA signature in predicting Recurrence-Free Survival (RFS), a multivariate Cox regression analysis was conducted, taking into account various covariates, including age at diagnosis, AJCC T stage, AJCC N stage, Gleason score, and pathological stage. Univariate analysis showed that all examined covariates had a significant association with RFS. However, even after accounting for these covariates, the risk score based on the 8-mRNA signature retained its prognostic impact on RFS. Therefore, the present study concludes that the 8-mRNA signature can be an independent prognostic factor for RFS. Furthermore, ROC analysis revealed that the 8-mRNA signature achieved an AUROC of 0.715 for predicting 2-year RFS and 0.747 for 5-year RFS. These values outperformed the level of detail provided by pathological staging alone. Pathological staging is a crucial risk assessment component associated with patient survival. Incorporating the 8-mRNA signature into risk assessment could help identify high-risk patients who might benefit from more aggressive treatment or additional adjuvant therapy. Additionally, the overexpression of SCUBE2 correlated with lower overall survival rates. At the same time, the other seven mRNAs showed significantly higher expression in the high-risk group than in the low-risk group. The functions of these seven mRNAs in prostate cancer remain largely unexplored. Based on the pathway analysis of GSEA KEGG, we speculate that these mRNAs may be involved in cell proliferation, cell cycle regulation, and immune response, possibly influencing prostate cancer immune escape or immunotherapy effects. Cell experiments further confirmed that the genes in the signature impact prostate cancer cell proliferation and invasion functions.
It is essential to acknowledge some limitations in our study. Firstly, the mechanisms underlying the predictive value of the identified mRNAs in prostate cancer remain to be thoroughly investigated. Additionally, the specific roles of the eight mRNAs in prostate cancer phenotypes require further clarification. Moreover, while the large sample size in the published dataset supports our findings, prospective testing in clinical trials and in vitro and in vivo studies is warranted to validate and further explore these observations.
The study developed a novel mRNA signature comprising eight mRNAs, enabling the prediction of survival outcomes in prostate cancer patients. Further investigations are required to understand the clinical implications and underlying mechanisms of these mRNAs, offering potential avenues for future research. These findings hold potential for advancing our understanding of prostate cancer biology and identifying therapeutic targets for improved patient care.
This research study utilized publicly available data from The Cancer Genome Atlas Program (TCGA). As such, no specific ethics approval or participant consent was required for this study. The data used in this research were previously collected and made available by TCGA following their own established ethical guidelines and data sharing policies.
As the data used in this study did not involve the collection or utilization of personal information or identifiable data from individuals, consent for publication was not required. The research was based on publicly available data TCGA and did not involve direct interaction or intervention with human participants.
All the analysis sample data in this paper come from the PRAD dataset of the TCGA database. Please refer to the Method section for the download website.
The work was supported by the grants of: Jiangsu Provincial Natural Science Fund (No. BK20210977).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Fanyu Peng and Min Wang improved and refined the project details and performed the experiments; Hao Zhang provided important recommendations and all tissue specimens; Xueyun Liu and Yesong Guo designed and supervised the study; Fanyu Peng prepared all figures and all tables. All authors reviewed and approved the manuscript.
We would like to acknowledge the TCGA for providing the necessary resources, facilities, and access to data that facilitated the smooth execution of this research. Their support has been essential in ensuring the successful completion of this project.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Peng F, Wang M, Zhang H, Liu X, Guo Y (2023) A Potential Panel of Eight‑mRNA’s Signature for Predicting Biochemical Recurrence‑Free Survival and Disease‑Free Survival in Prostate Cancer. J Clin Trials. 13:543.
Received: 05-Oct-2023, Manuscript No. JCTR-23-27321; Editor assigned: 09-Oct-2023, Pre QC No. JCTR-23-27321(PQ); Reviewed: 23-Oct-2023, QC No. JCTR-23-27321; Revised: 30-Oct-2023, Manuscript No. JCTR-23-27321(R); Published: 06-Nov-2023 , DOI: 10.35248/2167-0870.23.13.543
Copyright: © 2023 Peng F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.