Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Protocols - (2014) Volume 4, Issue 1
Background: The RIAS trial (Ramipril in Aortic Stenosis) is the first randomised, prospective, single centre, double blinded trial to examine the effects of Angiotensin Converting Enzyme (ACE) inhibition in asymptomatic Aortic Stenosis (AS).
Prognosis in AS depends on how the left ventricle responds to pressure overload; severe Left Ventricular Hypertrophy (LVH) carries an adverse prognosis. ACE inhibitors reduce LVH in other conditions and may have similar benefits in patients with AS. The aims of this study were three-fold:
1) To examine the regression of LV mass and changes in other LV physiological parameters using cardiac magnetic resonance (CMR)
2) To assess the safety and tolerability of ramipril in AS
3) To examine the potential improvement in exercise tolerance
Methods: 100 patients with asymptomatic moderate or severe AS will be enrolled. Patients will be randomized to placebo or ramipril (10 mg) for 12 months. At 0, 6 and 12 months, the patients will undergo a clinical assessment, phlebotomy, CMR scanning, echocardiography and a medically supervised Naughton protocol Exercise Test (ETT). Clinic checks at 2, 4, 12 and 14 weeks will be carried to titrate medication and monitor for adverse events.
Results: The primary endpoint of the trial is to measure change in LV mass. Secondary endpoints include changes in LV Ejection Fraction (LVEF), diastolic function parameters, perfusion, biochemical markers of LV function and exercise tolerance.
Conclusion: The RIAS trial is the first randomised, prospective, double blind trial to examine the effects of ACE inhibition in AS. If the trial is positive, our study would form the basis for a larger clinical outcome trial.
Trial registration: International Standard Randomised Controlled Trial Number 24616095
Aortic stenosis is common, occurring in 2% of the population over 65 years of age, rising to 7% in men over 85 years [1]. A transvalvular gradient of >50 mmHg is classed as severe [2] and when symptoms are present, the management is straightforward – valve replacement surgery is recommended [2]. The median survival without surgery in this group is 2-4 years depending on the symptoms [3,4]. For asymptomatic patients however, the management is less clear. Early valve replacement exposes patients to the risks of surgery and the ongoing risks of complications from prosthetic aortic valves for a longer period. Waiting for symptoms to occur, however, risks the development of Left Ventricular (LV) dysfunction and there is a small risk (~0.4% /year) of sudden death [5]. Currently, no medical therapy exists to prevent the progression of aortic stenosis or the onset of symptoms/ LV dysfunction. If such therapy existed, this would significantly aid the management of the asymptomatic group.
LV mass and function
As aortic stenosis progresses, intra-ventricular pressure rises and LV wall thickness (and thus LV mass) increase to normalise wall stress. This increase in LV mass may indicate the degree of compensation required by the left ventricle and thus also an indication of the development of dysfunction once decompensation occurs. The presence of symptoms (indicating decompensation) is not related to the valve gradient [6], but is related to LV mass, which is increased in symptomatic relative to asymptomatic patients [6,7] and correlates to the degree of LV dysfunction [7]. If this increase in LV mass could be reduced, it may be possible to delay the onset of symptoms and/or LV dysfunction and thus improve prognosis and/or delay the need for valve replacement. Angiotensin-Converting Enzyme (ACE) inhibitors are beneficial in LV dysfunction from other causes, both improving the dysfunction and reducing mortality [8,9]. They also reduce LV mass independently of the effect on blood pressure [10,11] and this reduction in LV mass appears to suggest an improved prognosis [12-14]. This effect of ACE inhibitors might also be true for aortic stenosis, but the theoretical danger of syncope caused by after load reduction has precluded the use of ACE inhibitors in this setting, and they are traditionally regarded as contra-indicated for moderate to severe aortic stenosis. However, there is no clinical data demonstrating adverse effects of ACE inhibitors in aortic stenosis; on the contrary, there is considerable basic science, animal and limited human data which suggests a beneficial effect of these drugs, which has been well reviewed by Routledge and Town end [15] and previously by Cox et al. [16].
ACE inhibitors thus have potential therapeutic benefits in aortic stenosis and a randomised prospective trial is necessary to examine what benefits ACE inhibition may bring to these patients. We propose a trial to examine in detail the physiological and function changes that may occur, and in particular whether ACE inhibitors may reduce LV mass.
Primary objective
To examine the regression of LV mass and changes in Left Ventricular (LV) physiological parameters using Cardiac Magnetic Resonance (CMR) in patients with moderate to severe aortic stenosis treated with the Angiotensin Converting Enzyme (ACE) inhibitor ramipril.
Secondary objectives
The secondary objective of the study is:
• To assess the safety and tolerability of ramipril in asymptomatic patients with moderate to severe AS.
• To examine potential improvement in effort tolerance as assessed by Exercise Treadmill Testing (ETT).
The study is a randomised, double-blind, placebo controlled trial of ramipril in asymptomatic patients with moderate to severe AS.
Primary endpoint
• Change in LV mass.
Secondary endpoints
• Change in left ventricular ejection fraction
• Change in other LV functional parameters assessed by MRI
• Change in biochemical markers of LV function (BNP)
• Time to symptoms of aortic stenosis or aortic valve replacement
• Change in distance walked & maximal effort tolerance on exercise treadmill testing
Study population
Subjects will be recruited from cardiology clinics at the John Radcliffe and surrounding hospitals. Inclusion criteria are as follow: all patients aged 18 years or over with moderate or severe asymptomatic AS by standard echocardiographic criteria (aortic valve area by continuity equation <1.5cm2, or valve gradient > 35 mmHg), with no other significant (> mild) valvular heart disease and an Ejection Fraction (EF) of > 40%. Exclusion criteria are: any contraindications for CMR scanning (eg: pacemakers, implantable defibrillators), pregnancy or terminal illness, serum creatinine >200μmol/L, Blood Pressure (BP) < 100/40 or > 200/110 mmHg, patients who had been prescribed ACEi or Angiotensin Receptor Blockers (ARB) over the previous 3 months or who were intolerant of ACEi or ARBs.
The study protocol was approved by the Oxfordshire research ethics committee C (reference 07/H606/139) and the Medicines and Healthcare products Regulatory Agency (clinical trial authorisation no. 21584/0226/001-004). All patients will be asked to give their written informed consent to participate in the trial. The trial was registered with the European Community clinical trials system (EudraCT no 2007-005224-32) and the ISRCTN register (24616095).
Study design
This will be a randomized, double-blind, placebo-controlled trial. After baseline visits, subjects will be randomized to receive ramipril or placebo for 52 weeks. The trial medication packs containing identical placebo and ramipril capsules will be prepared by a contract research organisation (Bilcare GCS, Crickhowell, Wales), and randomised using blocks of 4 with no stratification factors. Each pack will be identified by a sequential trial Personal Identification Number (PIN) and researchers will be blinded to the randomisation until after data analysis by the statisticians. The randomization schedule will be kept by the pharmacy at the John Radcliffe Hospital (for emergency code-breaks) and remain sealed until the end of the data analysis. Enrolled patients will be assigned medication packs in sequential order, corresponding to their trial PIN.
Subjects will be given an initial pack of study medication (containing ramipril 2.5 mg once daily or placebo) for a run-in period of 2 weeks to ensure no adverse symptoms occur. The ramipril will then be increased to 5mg daily at 2 weeks and again to 10mg daily at 12 weeks. Monitoring of symptoms and safety will be monitored by noting the occurrence of adverse events throughout the study, as well as documenting changes in laboratory parameters and vital signs at each visit. Monitoring visits will occur at 2, 4, 12, 14, 26 and 52 weeks. At weeks 26 and 52 all patients also underwent CMR scanning, echocardiography and exercise testing (Figure 1 and Table 1).
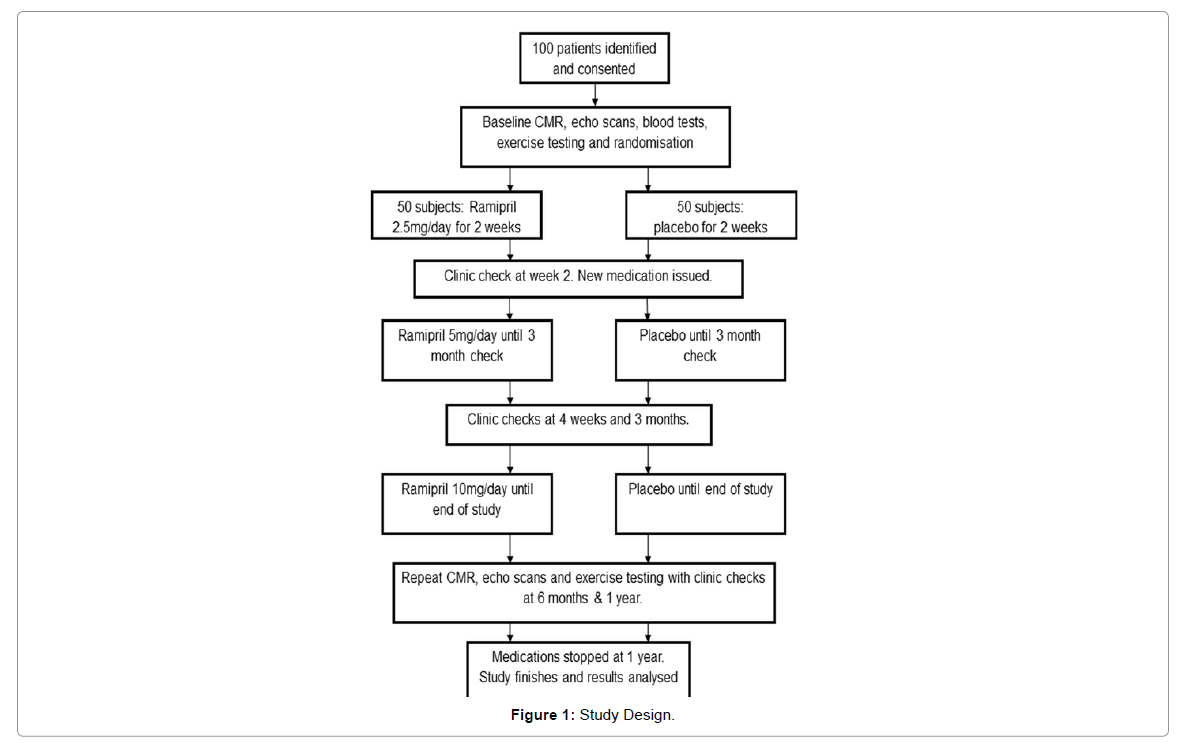
Figure 1: Study Design.
| Visit | Examinations carried out |
|---|---|
| 1 | Full Physical Examination Blood Tests (Electrolytes, Renal Function, Full Blood Count) Echocardiography Exercise Test Cardiac Magnetic Resonance Scan |
| 2 | Full Physical Examination Blood Tests (Electrolytes, Renal Function, Full Blood Count) |
| 3 | Full Physical Examination Blood Tests (Electrolytes, Renal Function, Full Blood Count) |
| 4 | Full Physical Examination Blood Tests (Electrolytes, Renal Function, Full Blood Count) |
| 5 | Full Physical Examination Blood Tests (Electrolytes, Renal Function, Full Blood Count) |
| 6 | Full Physical Examination Blood Tests (Electrolytes, Renal Function, Full Blood Count) Echocardiography Exercise Test Cardiac Magnetic Resonance Scan |
| 7 | Full Physical Examination Blood Tests (Electrolytes, Renal Function, Full Blood Count) Echocardiography Exercise Test Cardiac Magnetic Resonance Scan |
Table 1: Visit Summarises.
Baseline assessment (Visit 1)
Demographics: The date of birth and gender will be recorded.
Medical History: A medical history will be taken to elicit any major previous medical problems and patients will be asked to bring their current medication with them. Where possible an attempt will be made to establish the aetiology of aortic stenosis, i.e. congenital, rheumatic, degenerative. Specifically, the presence or absence of the following symptoms will be ascertained:
• Shortness of breath (including NYHA functional class).
• Chest pain.
• Syncope.
Physical examination: Height, and weight will be recorded and body mass index calculated. Resting pulse and Blood Pressure (BP) measurements will be measured after the patient has been seated for at least five minutes. Blood pressure will be recorded in a seated position and after standing for 2 minutes. A clinical examination will be conducted, including auscultation of the chest, examination of the abdomen and assessment of any ankle oedema.
Resting ECG: A 12-lead ECG will be taken for each patient. The following ECG parameters will be recorded: Heart Rate (HR), rhythm, any degree of atrioventricular block and the presence of left ventricular hypertrophy using the Sokolow-Lyon criteria.
Clinical laboratory sampling: Venous blood samples will be taken for analysis of full blood count (FBC), Urea and Electrolytes (U & E) including creatinine, Liver Function Tests (LFTs), Total Cholesterol (TC) and B-type Natriuretic Peptide (BNP). Patients with a documented U&E result in the previous 6 months will have their Glomerular Filtration Rate (GFR) calculated from the creatinine level to ensure their renal function meets the inclusion criteria.
Echocardiography protocol
Transthoracic echocardiography will be carried out using a Philips iE33 advanced echo system (Philips Medical Systems, Best, Netherlands) equipped with 2.5-3.5 MHz transducers.
A full echo study will be performed according American Society for Echocardiography guidelines for assessment and classification of AS and chamber quantification [17]. Left ventricular volumes and ejection fraction will be calculated by Simpson’s rule. LV diastolic function will be assessed using tissue Doppler in line with the American Society of Echocardiography guidelines [18].
Cardiovascular magnetic resonance protocol
The comprehensive CMR protocol will be carried out at baseline and at 12 months. At 6 months, only LV volumes, mass and function will be assessed. Patients will be scanned using a 1.5 Tesla Avanto CMR system (Siemens, Erlangen, Germany). Left ventricular mass, volumes and function will be assessed using a stack of steady state free precession short axis cine images of the LV, acquired using breath-holding and cardiac gating in accordance with the Society for Cardiovascular Magnetic Resonance (SCMR) guidelines [19]. LV mass, volumes and function will be analysed using Qmass software (Medis, Leiden, Netherlands) using semi-automatically applied endo and epicardial contours to short axis slices in end-systole and end-diastole. LV mass will be calculated from the end-diastolic myocardial volume based on known myocardial density (1.05 g/cm3), and all parameters will be indexed to body surface area.
The aortic valve will be assessed using Steady State Free Precession (SSFP) cine sequences acquired in a short axis slice at the aortic valve tips in mid-systole. Breath-hold in-plane and through-plane phase contrast velocity mapping will be carried out just distal to the aortic valve (at the vena contracta) to determine the peak velocity across the aortic valve. Planimetry of the aortic valve area and velocity analysis will be carried out using Argus software (Siemens Medical Solutions, Erlangen, Germany) [19].
Myocardial strain: A gradient echo-based tagging pulse sequence will be performed in the long axis (four chamber) and in the basal, mid ventricular and apical short-axis slices with a segmented k-space, multi-shot sequence. Spatial modulation of magnetization [20] produced images with a grid-based pattern of horizontally and vertically modulated ‘stripes’ 1cm apart, will be acquired during a single breath hold. The temporal resolution was < 40 ms in all data sets, and 15-25 frames per cardiac cycle will be recorded, depending on heart rate. Semi-automated analysis will be performed using CIM software (CIMTag2D v.7, Auckland MRI Research Group, New Zealand) by aligning a grid to the myocardial tagging planes in end diastole. Longitudinal Strain (LS) and Strain Rate (SR) will be calculated from the long axis (4 chamber view) images, Circumferential Strain (CS) and SR from the left ventricular short axis images.
Assessment of interstitial myocardial fibrosis using T1 mapping
T1 mapping will be performed using a Short Modified Look Locker Inversion recovery (ShMOLLI) technique, applied to a single midventricular slice in the short axis position. The image will be manually contoured to outline the endo- and epicardium, and the average T1 value over this whole slice calculated using in-house software (MyoCardial Regions Of Interest) MC-ROI in IDL (Interface Definition Language) v.6.1 (ITT Exelis, Mclean, Virginia, US) as previously described [21]. This technique was previously validated by our group as providing a measure of cardiac interstitial fibrosis in AS [21].
Myocardial perfusion and late gadolinium imaging
Myocardial perfusion reserve will be assessed following administration of adenosine at a rate of 140 μg/kg/min for 3 minutes before perfusion imaging. Gadolinium-based contrast (gadodiamide [Omniscan]; Nycomed Amersham, Little Chalfont, UK) will be administered intravenously as a bolus at a dose of 0.03 mmol/kg (injection rate 6 mL/s), to maintain a linear relationship between signal intensity and absolute perfusion. The gadolinium was followed by a saline flush of 15 mL at the same rate. Perfusion imaging will be performed at every heartbeat during the first pass of the contrast bolus with the use of a T1-weighted fast (spoiled) gradient echo sequence with saturation-recovery magnetisation preparation, as described previously [22]. Perfusion images will be acquired in 3 short-axis sections at basal (between LV outflow tract and the papillary muscles), mid-ventricular, and apical levels chosen according to guidelines [23]. To allow sufficient contrast washout, rest perfusion imaging will be performed at least 20 minutes after the stress study. The perfusion images will be processed by tracing endocardial and epicardial contours (QMass software, version 7.2, Medis, Netherlands). A region of interest will be drawn in the LV blood pool, avoiding any papillary muscles therein, to permit the derivation of an arterial input function. The myocardium will be divided into equiangular segments based on the American Heart Association segmentation model [23]. Rest and stress myocardial perfusion upslopes will be calculated using 5 point linear fit model of signal intensity vs. time and normalized to the LV blood pool upslope [24]. Myocardial perfusion reserve index (MPRI) will be derived for each of the 16 segments per subject, defined as the ratio of stress to rest normalized myocardial perfusion upslopes. The average MPRI of the 16 segment value was used.
Late Gadolinium Enhancement (LGE) images will be acquired after an additional 10 minute delay with the use of an inversion-recovery prepared, segmented gradient echo sequence, as previously described [25]. In all patients, imaging will be repeated for each short-axis image in 2 separate phase-encoding directions to exclude artifact. Mid-wall LGE will only deemed to be present when the area of signal enhancement could be seen in both phase-swapped images and in a cross-cut long-axis image by the independent observers.
Exercise treadmill test
An exercise treadmill test will be carried out according to standard procedures and will be medically supervised. A linear graded exercise protocol (Naughton) will be used to assess exercise capacity as a continuous variable, and patients will be asked to continue walking until they can walk no further. This test may be performed within 2 weeks of the relevant clinic visit, though baseline testing will be performed prior to commencement of the trial medication.
Visit information
Visits 2-5: The patient’s medical status will be assessed and any adverse events or changes in prescribed medication will be recorded. Patients will be asked to bring all their study medication with them and a pill count will be undertaken to assess compliance. Patients will be asked about any new symptoms and specifically cough, dizziness, syncope, breathlessness, tongue or throat swelling and skin rashes. In addition they will undergo a brief physical examination to check the pulse (rate and rhythm), blood pressure (seated and after 2 minutes standing) and to check for signs of fluid retention (raised JVP, basal lung crepitations, S3 gallop, hepatomegaly, ascites, sacral or leg oedema) that could indicate underlying heart or renal failure. A venous blood sample will be taken for U&E measurements to monitor renal function and serum potassium level. Patients will then be issued with new randomisation packs containing either placebo or ramipril 5mg once daily for 3 months at visit 2 and 10 mg once a day at visit 4.
Visit 6: This visit will take place 6 months after randomisation. During this visit the same assessment as for visit 2 will be undertaken. In addition, imaging and physiological monitoring as performed in visit 1 will be performed. Patients will be issued with new randomisation packs containing either placebo or ramipril 10mg each once a day for 6 months.
Visit 7: This visit will take place 12 months from randomisation. At this visit patients will undergo the same procedures as at baseline (visit 1). At the end of the procedures, study medication will cease.
Post study telephone check (7-10 days after visit 6)
There will be a telephone call made to the patient 7-10 days after cessation of study medication. The purpose of this call will be to ensure that patients are stable after withdrawal of study medication. Patients will be asked about their general health, any new symptoms and any change in their exercise capacity.
Minimisation of bias
A contract research organisation will be commissioned to implement the randomisation process and provide the medication packs. Both the ramipril and placebo will be formulated and supplied in identical capsules sealed in identical medication packs. The allocation to placebo/ramipril will be randomly assigned and the medication packs will be consecutively numbered with the trial PIN. Sealed randomisation envelopes will be kept unopened and in a secure place by the pharmacy at the John Radcliffe Hospital and the Investigators. The medication packs will be kept in the John Radcliffe Hospital pharmacy which will dispense them to trial participants. Patients will be enrolled and assigned a PIN consecutively, and issued with the medication pack corresponding to the PIN. Both trial patients and investigators will be blinded to the nature of the study medication dispensed and all image and spectroscopic analysis will be conducted while blinded to study treatment. Once all collected data has been analysed, the randomisation code will be broken by the study investigators and the medication data entered into the analysis. Should urgent un-blinding of a trial patient’s medication be required (when requested by a clinician treating the patient), this will be provided by the pharmacy at the John Radcliffe Hospital. This is normally a working-hours service, but in exceptional circumstances could operate at any time. The trial investigators have reviewed the clinical safety of the study and do not feel that a 24-hour un-blinding service would be required for the appropriate treatment of patients, either within the John Radcliffe Hospital or elsewhere.
Definition of end of trial
The end of trial is the date of the last follow up contact of the last patient.
Withdrawal of patients/subjects
Patients may withdraw or be withdrawn from the study for any of the following reasons:
• Patient decides not to continue with the study.
• Pregnancy.
• Requirement for ACE inhibitor or ARB medication on clinical grounds as assessed by the patient’s physician
• Administrative decision by the Investigator.
• Development of significant hypotension (sustained systolic BP <90mmHg if asymptomatic or <100mmHg if symptomatic; or sustained diastolic BP<40mmHg regardless of symptoms)
• GFR< 30mls/minute or creatinine level rising to > 50% above baseline level. If GFR falls to between 30-59mls/minute, patients may remain in the study but would not receive gadolinium contrast during the CMR scans
• Significant protocol deviation.
• Patient non-compliance with treatment regime or study requirements.
• An adverse event which requires discontinuation of the study medication or results in inability to continue to comply with study procedures. Under normal circumstances, a SUSAR will result in unblinding of the data for that patient and withdrawal from the study.
• Disease progression which requires discontinuation of the study medication or results in inability to continue to comply with study procedures.
Concomitant medication
Throughout the study, prescription and dose adjustment of concomitant medications other than ACE inhibitors or ARBs will be left to the discretion of the primary physician. Any prescribed, regular medication, other than the study medication taken during the study, will be recorded in the CRF.
Assessment of safety
Serious adverse events which may be due to disease progression include hospitalisation for:
• Heart failure
• Syncope
• Angina
• Aortic valve replacement
In the case of heart failure, syncope and angina, it may be difficult to be certain whether the event was due to disease progression or related to the study medication. These would therefore be treated as serious adverse events. Where disease progression was felt to be the very likely cause (including aortic valve replacement), this will be recorded in the CRF but would not require immediate reporting.
All adverse and serious adverse events will be reported.
Statistical analysis methods
Descriptive analyses: For each study measure descriptive statistics within each randomised group will be presented at each time point (baseline, 6, 12 months).
Statistical analysis of primary and continuous secondary outcomes: Primary and continuous secondary outcomes will be analysed using a linear regression model adjusting for baseline values. Results will be presented as adjusted mean difference in change in LV mass (g) between randomised groups at 12 months with 95% Confidence Intervals (CI) and associated two-sided p value.
Statistical analysis of time to event outcomes: Time to event secondary outcomes (time to death, time to AVR) will be presented using Kaplan-Meier curves and differences between groups assessed using the log-rank test. Hazard ratios and associated 95% confidence intervals will be computed using proportional hazards regression.
Sample size
Data on human subjects are limited in this field. The power calculation is based on changes in LV mass, for which there are preexisting data, indicating a likely effect of ACE inhibitors, and baseline CMR measurements of LV mass in patients with aortic stenosis. The mean baseline LV mass in a recent study of aortic stenosis using CMR was 142 g/m2 with a standard deviation of 35 g/m2. The mean reduction in LV mass with ACE inhibitors from a large meta-analysis11 of antihypertensive treatments was 15%, which equates to 21.3 g/ m2. Using these figures, and a one-sided paired test (only including reduction in LV mass) with 85% power (β error) and 95% confidence (α error), the number needed in each study group is 43. By including 50 in each group, we will allow for a 15% drop-out rate.
Level of statistical significance
A p value of 0.05 or less will be considered statistically significant. No adjustments will be made for multiple testing.
Criteria for the termination of the trial
The trial will be terminated prematurely if any safety concerns regarding the treatment under scrutiny arise.
Procedure for accounting for missing, unused, and spurious data
All attempts will be made to ensure capture of all relevant study data. Data checks will be carried out to find values that are implausible or out of range. These values will be queried, and corrected if possible. If values are thought to be spurious and it is not possible to correct them that data will be disregarded for the purposes of analysis. If missing data is substantial then multiple imputations will be used to assess the impact of missing data in a sensitivity analysis.
Statistical analysis plan
Prior to analysis a statistical analysis plan will be written, detailing the proposed analysis of primary and secondary objectives for the study. Any deviation from this plan will be described and justified in the final report.
Analysis population
Primary and secondary analyses will be conducted on the Modified Intention to Treat (mITT) population. This will include all participants who were randomized and who receive study medication and from whom at least one efficacy measurement is obtained after first treatment with study medication. All patients will continue to be followed up clinically according to their treating physician’s plan after the end of the study, and clinical follow-up data may be incorporated into subsequent analyses.
The RIAS trial is the first randomised, prospective, double blinded trial to test that hypothesis that ACE inhibition in AS can induce positive remodelling of the left ventricle using CMR endpoints. CMR will allow us to study the physiological response of the left ventricle to ACE inhibitors in great detail, including an assessment of the effect of ACE inhibition on left ventricular perfusion, interstitial fibrosis, strain and diastolic function. If this trial is positive, this might translate into improved clinical outcome in patients with asymptomatic AS and could potentially delay the need for AVR in the future. This study would then form the basis for a multi-centre trial which would be carried out over a longer period and would be powered for clinical outcomes to determine this.