Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2024)Volume 17, Issue 2
The primary objective of the target-decoy strategy is to estimate the False Discovery Rate (FDR) for reported peptide or protein matches during a protein database search. Various strategies, such as decoy database generation methods and search engine combinations, have been explored. Earlier research investigated the influence of decoy construction models and showed stochastic/statistical methods to be more promising and accurate than basic sequence reversal or shuffling methods.
In this paper, we propose a novel decoy creation framework based on proteins’ significant biological signatures, patterns, and profiles, using stochastic models such as Markov. As part of the proposed approach’s flexibility, decoy sequence generation can be adapted to digestion sites and be peptide-based or protein-based.
For comparison and benchmark purposes, we investigated a standard MS/MS data set of two well-known protein pools based on E. coli peptide fragments to compare the proposed approach to standard methods by assessing the false discovery rate and identification correctness.
When compared to default methods, the false discovery rate was quite high. The imbalanced number of discovered patterns in the two pools has resulted in an improved accuracy and specificity for sequences with the most signatures. For certain examined samples, the proposed method improved the correct and incorrect identification ratios by 12.3 percent and 7.7 percent, respectively.
Protein identification; E. coli ; Peptide fragments; Flexibility
In general, protein motifs or patterns sequences are conserved amino acid sequence patterns with biological significance and are a collection of biologically relevant signatures seen in a single protein linked to biological information about the protein family, domain, or functional location identified by the signature. Motifs are found in protein-protein interactions, protein-nucleic acid interactions, post-translational modification, protein transport, signal transduction and other biological processes. Biological data on motifs and patterns can be acquired in both general and specialized databases. General databases provide an exhaustive collection of relevant data and a rather diverse set of biological information. Specialized databases, on the other hand, are more well-built and homogeneous. Available tools enable the online scan of databases, but they are powerful and time-consuming.
The identification, quantification, and subsequent biological interpretation of peptides and proteins is solely reliant on matching fragment ion spectra to peptide sequences. Target decoy database searching is now the de facto standard approach, with each peptide spectrum match given a false discovery rate estimate [1]. While several mathematical models and strategies for decoy database generation have been developed to enhance identification, none of these approaches incorporate information about protein motifs and patterns.
In this paper, we propose an alternative decoy approach considering these motifs and patterns that goes beyond peptide or protein sequence presence to inform on the presence of protein family domains and functions. We estimate a false discovery rate as a function of protein signatures in this framework, which may be expanded to a wide variety of possible protein information [2].
The proposed approach begins by searching the target database for motifs and patterns in each protein. Known signatures are queried from public databases such as ExPASy, Prosite, NCBI, Pfam, and InterPro [3-7].
Compiled patterns are merged and stored in a tabular structured format, and then we programmatically map each protein in the target database to its set of motifs and proceed into the decoy sequence generation process (Figure 1).

Figure 1: Pattern/motif decoy-based generation approach. Classical methods consider the whole target protein sequence while the proposed approach singles out and generates decoys for each pattern.
Mixing classical decoy generation methods with the proposed framework would have a poor impact on the false discovery rate (in reverse and random decoy methods, symmetric and amino acid-rich proteins are unaffected), and thus stochastic/statistical models that fit the patterns’ structure, such as the Markov model, are required.
Markov chain and hidden Markov models are stochastic/ probabilistic models that attempt to capture the behaviour of an event by computing the probabilities of transitions between its states over time and are the new trend in bioinformatics for accurately describing many biological processes, drug effectiveness and disease progression [8,9]. While a Markov model assumes that the current observation will only be dependent on the most immediate previous one, Hidden Markov Model (HMM) is a double stochastic process having an underlying stochastic process that is neither observable nor quantifiable, but can only be witnessed via another set of stochastic processes that form the sequence of observed symbols, or amino acids in the current context. In our approach, the underlying graph of the fitted model represents the structure or the anatomy of the reversed protein’s motif sequence.
For benchmark purposes, a standard Liquid Chromatography- Mass Spectrometry/Mass Spectrometry (LC-MS/MS) spectra created by scanning the Human Proteome Atlas (HPA) project’s PrEST sequences for 191 overlapping pairs of PrEST sequences were investigated [10,11]. Two pools, A and B, were formed from these pairings. Each pool included just one of each pair’s PrESTs. Pool A+B was formed by combining pools A and B to create a third pool. Each pool was mixed into a background of a tryptic digest of 100 ng Escherichia coli (BL21(DE3) strain), resulting in three mixtures, Mixture A, Mixture B and Mixture A+B. An equal amount of each mixture was analysed in triplicate by LC- MS/MS in random order. Full metadata can be found under the Pride database [12].
Search engine choice and accuracy were not part of this work’s main matters, and we are comparing the proposed method to classical decoys. Crux-Tide is used for searching mixtures spectra against a well-prepared target database. The protein level false discovery rate is estimated using Crux-Percolator [13-15]. Correct identification performances are compared at an FDR threshold of 1%.
In general, all models performed well and detected a considerable percentage of proteins in the mixtures. For instance, Figure 2 shows a circular bar-plot draws correct and incorrect identification ratios by sample, pool and decoy method (Figure 2).

Figure 2: Circular bar plots for correct and incorrect protein identifications by pool and sample. Correct identifications are indicated in blue, while incorrect identifications are indicated in red.
Even if some proteins are missing or wrongly identified due to background noise, the identification still yields a high proportion of PrEST proteins. In Figures 3A and 3B, when we examine the false discovery rate distribution across samples, we see that the relatively low FDR distribution for blank samples has led to previously witnessed erroneous detection, and that samples A, B and C share a similar distribution shape. Pool A (sample C) has a slightly lower FDR than other samples, which is explained by differences between the two pools in terms of proteins lengths and number of detected patterns (Figures 3A and 3B).

Figure 3: Pools’ motifs/patterns and proteins length distributions.
(A): Motifs/patterns by position and pool; (B): Violin distributions
of proteins length in pool A and pool B. 
There have been earlier theoretical predictions about probable performances, but a solid review would make more sense. The three benchmarked decoy models can be compared in a multitude of ways.
First, in terms of computation complexity, as protein sequences are screened for signatures using database queries, our proposed method has a large and exhaustive search stage. This time-consuming process is executed once for each target protein database. The compiled motif set can be updated, so there is no need to redo the entire operation. On the other hand, the other models do not require any pre-tasks.
Second, our method produces fewer false positives. A protein that is not present in a sample but is nevertheless reported by the search process is referred to as a false identification. As shown in Figure 2, the reverse decoy performs badly and has the highest false identification. Third, in terms of correct identifications, our decoy model successfully identified more than 91% proteins in each pool. The other models fared worse, with an incorrect detection ration of 84% in the case of the reverse decoy in sample A. In Figures 4A and 4B, when compared to shuffle and reverse, the proposed method had a greater false discovery rate and was more selective at higher FDR levels (Figures 4A and 4B).

Figure 4: Number of accepted decoy and target proteins vs. FDR. (A):
Decoys vs. FDR; (B): Targets vs. FDR. 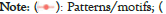
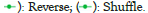
Figure 5 shows that there is a strong correlation between the number of accepted decoys and targets in our approach (A factor of 0.93 with an adjusted R of 0.97). The motif-based decoy clearly respects Target-Decoy Approaches (TDAs) fundamental principle that false matches are spread evenly throughout the target and decoy data-bases, and thus would be appropriate and straightforwardly adapted for multi-stage searches to allow an unbiased TDA-based FDR estimation (Figure 5).

Figure 5: Accepted targets proteins vs. decoys proteins. 
Finally, the impact of protein lengths and the number of patterns on the outcomes of identifications can be seen in Figure 2 in samples B (Pool B) and C (Pool A). More patterns in sample C led to fewer false positives. Because of short protein lengths and few biological patterns, we accurately assigned more proteins in pool B (more than 97%) at the same FDR threshold resulted.
Formulating a novel method that may increase accuracy is not a chancy process. Our contribution provides a framework for decoy creation that outperforms current models, as well as a compelling argument for delving further into models that do not simply regard protein as a linear sequence of amino acids. Biological information, when supplied or incorporated during the decoy model building process, can improve separation and identification power.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Bensellak T, Moussa A (2024) A Protein Signatures-Based Decoy Framework for Improved Protein Identification in Target-Decoy Search Approach. J Proteomics Bioinform.17:667
Received: 25-Jul-2023, Manuscript No. JPB-23-25084; Editor assigned: 27-Jul-2023, Pre QC No. JPB-23-25084 (PQ); Reviewed: 10-Aug-2023, QC No. JPB-23-25084; Revised: 17-Aug-2023, Manuscript No. JPB-23-25084 (R); Published: 24-Jun-2024 , DOI: 10.35248/0974-276X.24.17.667
Copyright: © 2024 Bensellak T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.