Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Review Article - (2015) Volume 8, Issue 3
Proteomics is the study of proteins on a genome-wide scale. Within the wide field of functional OMICS, proteomics has become a useful tool. The completion of genome sequencing projects and the improvement of methods for protein characterization surges this action forward. Presently, the usage of proteomics is being extended to analyze the different features of proteins including the activities and structures, and protein-protein interactions. Proteomics research is quite advanced in animals, yeast and bacteria, but it is still in the beginning stages of plant research, due to its highly complex and dynamic status. In view of the advances in crop biotechnology, it is critical to understand the role of proteins during plant development and response to biotic and abiotic stimuli. In this review, we presented several plant proteomic studies to illustrate the applications of proteomic studies in crop productivity. The advances in proteomics in recent years include protein isolation methods, mass spectrometry, protein-protein interactions and post translational modifications. We further discuss the strengths and weaknesses of proteomic technologies and the limitations of current techniques in the perspective of plant biology. We conclude that advances in protein interactions and bioinformatics will have an increasing impact on better understanding the various functional aspects in plants, such as PTM, subcellular localization and protein interactions.
Keywords: Proteomics, Bioinformatics, Omics, Genomics, Organelle proteome, Post translation modification, Protein-protein interaction
Proteomics involves the study of the protein complement of the genome [1]. Plant proteomic projects include structural proteomics of the whole organism, organs, tissues, cells, and sub cellular compartments, as well as comparative proteomics on various processes. Yields of crops are reduced by numerous abiotic and biotic factors, such as flooding, drought, salinity, acidity, and nutrient limitation. Plant interactions with other organisms trigger biotic stresses and defenses. Research has greatly increased in the past decade not only by observing the existence of each process, but their interactions as well. Recent studies have found that responses to abiotic and biotic stresses influence each other both positively and negatively [2].
Due to the various environmental changes in the last few decades, it has become more evident that in the event of drastic shifts in the environment, fresh crops will more than likely not be available to use as a source of food [3]. Therefore, several alternative techniques, such as molecular markers, association mapping, candidate gene sequencing, and whole genome scanning, along with gene networks, and allele mining, have been adopted to sustain the productivity and increase crop yield. Markers are developed from expressed sequence tags; those developed from gene sequence data are called functional markers. Functional marker research has been developed uniquely around plant species, and is used more than random markers, because it is completely linked to the desired, or studied, allele.
A major challenge in the field of biotechnology, however, is the gap between the rate of development of new technologies and the development in applied breeding programs for crop improvements. The complete genome sequences of model plants, Arabidopsis [4], and rice [5] provide insight into several aspects of plant biology. However, functional genomics studies on the majority of crop plants are still in their early stages because some species have genome duplications, self-incompatibilities, and a long generation time. In such cases, the proteomics approach is a powerful tool for analyzing the functions of the plant genes or proteins. Genome-sequence data and inferred protein-sequence data can be used to identify proteins and to follow sequential changes in protein expression in an organism. Currently, we are capable of developing genotypes by finding and matching the right genes that can perform better in harsh environments [6].
Gene-to-metabolite networks are typically constructed using multivariate analysis or data mining [7]. Gene regulatory networks describe how genes interact, and it incorporates post-transcriptional events such as protein targeting and covalent protein modification [8,9]. Some DNA families, for example, contain kinase protein and the detoxification proteins of glutathione S-transfer gene (GST) family and are made up of over 100 distinctive proteins. However, ironically, outside the DNA family, these proteins vary significantly [10]. Gene families are often repetitive due to mutations and duplications. However, the knowledge is still limited on how these genes in a certain family may be better observed given that more than one family is performing the same task.
The creation of modern systems biology comes from the need to gather information from genome-scale studies and being able to present them in biological interpretations. Systems biology is continuous, though some researchers tend to define systems biology as utilizing dynamic modeling or multidimensional data analysis [11]. Therefore, development of methods for protein characterization is critical to complete functional genomics.
Even though high-resolution LC-MS (Liquid chromatography– mass spectrometry) strategies have resolved some of the issues with direct analysis of complex peptide mixtures, other problems have risen from metabolites and proteases in great abundance -e.g. cell wall polysaccharides and polyphenols, lipids, and starch [12]. Therefore, these substances were heavily diluted or removed completely. However, such purification can often lead to sample loss [13]. Hence, the use of self-packed nano-LC columns for purification before matrix assisted laser desorption/ionization Mass Spectrometry (MALDI-TOF MS) has been employed successfully [14].
Protein extraction
The protein-pellet homogenization method and the lyses buffer have a significant influence on protein solubilization and separation in classical proteomic analysis of plants. Most of the protocols used in extraction of proteins from plant tissues are based on either phenol or TCA/acetone precipitation methods. The TCA/acetone procedure has some limitations. For example, the resulting pellet may be hard to dissolve; nucleic acids longer than ~20 nucleotides can be precipitated; and in some cases, proteins can be hydrolyzed by TCA. Protein extraction and sample preparation, particularly from recalcitrant tissues such as grape leaf, cucurbit pericarp, pine needle (generally containing high level of interfering substances, such as phenolic compounds, proteolytic and oxidative enzymes, terpenes, organic acids and carbohydrates) are among the most challenging aspects of proteomic analyses [15].
It has been demonstrated in recalcitrant plant tissues and others that TCA-acetone–phenol-based method is more effective than TCA/acetone precipitation alone prior to protein solubilization [12]. However, various protein extraction and solubilization buffers with a diversity of chemical compositions and concentrations have been used, such as Mg/NP-40 buffer and Tris Buffer. The use of PMSF, a protein inhibitor, and many other protease inhibitors, function to minimize the proteolysis of proteins during isolation [16].
Protein separation
Separation of complex mixtures of proteins is carried out by methods including analysis of the pI, Mr, solubility, and the relative abundance. 2-DE, which encompasses IEF from the first dimension and SDS-PAGE from the second dimension, allows this separation to occur. In addition to enabling the separation of complex mixtures of proteins, 2-DE also consents for proteins that have undergone PTMs to be further isolated for structural analyses using a mass spectrometer. The MALDI-TOF MS, MS alone, electrospray ionization (ESI) or Edman micro sequencing is used by the mass spectrometer to analyze the structures of the proteins [17].
In order, 2-DE MS comprises of, sample preparation and protein solubilization, protein separation protein detection and quantification, computer assisted analysis of 2-DE patterns, protein identification and characterization, and 2-D protein database construction. When studying protein spots on a 2-DE gel in depth, the resolution is often restricted due to aspects such as size, abundance hinder by denaturation and intermolecular interaction, and other electrophoretic properties [18]. Therefore, to refine the resolution and sensitivity, it has been found helpful to divide the whole proteome into discrete parts, including organelles, sub cellular compartments, and multi protein complexes.
LC-MS-based qualitative analysis of plant systems
A completely sequenced genome significantly aids in the process of obtaining results from proteomic research. However, only a few eukaryotic organisms’ genomes have been sequenced [19]. There have been 1,792 eukaryotic genomes sequence dpaling in comparison to the 29,369 sequenced prokaryotic genomes used to study evolutionary processes, such as lateral gene transfer [20]. In contrast, other genomic research has been focused on model organisms such as the human, rat, drosophila, A. thaliana, and rice. Recent research has brought forth label-free studies of the plant proteome. However, label-free techniques are not without problems, particularly when concerning plant samples containing high abundance proteins [19] and large amounts of coeluting peptides during LC-separation –e.g. in cell lysates [21].
Biological Mass spectrometry based quantification
Biological mass spectrometry (MS) is the use of a mass spectrometer in proteomics, which involves proteins (now peptides in this stage of the process) that have been separated by SDS-PAGE. These are then introduced to the mass spectrometer via MALDI or ESI. After being introduced to the spectrometer, the mass-to-charge (m/z) of peptides are measured generating MS spectra [22]. Peptides are commonly fragmented by collision (induced dissociation), which fragments the ions of peptides. MS-based quantification of proteins relies on two strategies: label free and stable isotope labeling [23,24]. Label-free protein quantification is split into techniques relying on either the intensity of the peptide ion in the mass spectrometer or the number of scans, the latter requiring too much data to be efficient for PTMs characterization. Stable isotope labeling is characterized by chemical labeling (relying on sulfhydryl groups of cysteine, free amines), or by enzymatic labeling performed on intact plant tissues or liquid cultures. Plants are often labeled with 15N along with K15NO3 as the only nitrogen source during growth in soil or in cell suspension cultures enabling a large quantity of peptides enriched in 15N to be identified, 98% of which will be incorporated. An alternative method called SILAC (Stable Isotope Labeling of Amino acids in Cell culture) is used, in which the cells are supplied with labeled amino acids, instead of 15N, yielding 80% of the quantified proteins incorporated, because plant cells already synthesize amino acids [25]. Therefore, a number of stable isotope labeling techniques using 2H, 13C, 15N, or 18O isotopes were developed for MS-based quantitative proteomics [26-28].
Multidimensional protein identification technology (MudPIT) uses a combination of liquid chromatography (LC-MS/MS) and high performance liquid chromatography (HPLC), enhances the separation of peptide mixtures using strong cationic exchange and 2D liquid chromatography [29,30]. Thus, by improving sample separation methods along with efficient techniques of peptide mixture separation, the overall analysis of proteome can be enhanced to identify proteins of all functional and physical classes, which are found in low abundance.
Several attempts have been made to analyze the differential proteome of crop plants in response to various stresses, including toxic abiotic and biotic factors, such as metals [31], salinity [32], flooding [33], ultraviolet-B radiation [34] and pathogen infestation. The proteomic approach has also been applied to unravel the expression of allergens in transgenic plants [35] and in comparing allergens between cultivated and wild types [36].
Abiotic factors
Abiotic stressors affect the way a plant grows and performs various functions.
Metal toxicity: Toxic metals, such as Cd, Cu and Al, are major pollutants in soils and their levels are rapidly increasing within the environment. They are frequently taken up by plant roots, thereby severely interrupting several physiological and biochemical pathways, and thus restrict growth and development, ultimately leading to cell death. To investigate the Cd-responsive proteins in soybean, suspensioncultured cells were exposed to various concentrations of cadmium and labeled with 35S-methionine for three different time periods [37]. Significant up-regulation of first-line anti-oxidative genes, such as superoxide dismutase, have also been identified in other proteomic analyses of stress responses to several metals, including cadmium [38].
Flooding stress: Among various crops, soybean, rice, maize and tomato are considered too highly sensitive to flooding stress. Various studies on hypoxia or waterlogging stress have been carried out using cytosolic and membrane proteins from roots and have shown regulation of cytosolic ascorbate peroxidase-2 under flooding stress in soybean [39-41].
Chill stress: In the process of understanding the root of chilling stress responses, it is found that the exploration of root proteome appearance and certain proteins may be critical. A group of novel membrane stability related proteins were up regulated during chilling stress in peach fruit and were identified using electrospray ionization MALDI-TOF MS [42]. These proteins are believed to be significant in our understanding of the stress responses in plants, the reason being that they participate in many cellular processes, including detoxification, vesicular trafficking, and metabolism [43].
Heat and water stress: High temperature is detrimental to the growth of cool-season plant species in many temperate areas or in subtropical regions. Heat stress can cause changes in various metabolic processes, such as protein denaturation, inhibition of synthesis of normal cellular proteins, and induction of some heat shock proteins (HSPs). The HSPs function in the stabilization of proteins, while membranes assist in protein refolding under stressful conditions [44].
Water stress is the most commonly occurring abiotic stress, particularly in rain fed crops, such as peanut, maize, and sorghum. Proteome analysis of the peanut leaf was conducted using twodimensional gel electrophoresis in combination with sequence identification using MALDI-TOF to determine their identity and function related to growth, development, and responses to stresses. The 2DE reference map was derived from a drought-tolerant peanut leaf tissue [45] and serves as the basis for further investigations of peanut physiology, such as detection of expressed changes due to biotic and abiotic stresses, and plant development.
Protein response in plants has proved to vary greatly, however, it is most common for proteins in response to oxidative damage arise in abundance in roots [46]. Analysis of proteins in soybean leaf by oxygenisotope- fractionation revealed a 32% mitochondrial ATP synthesis decrease inhibiting photosynthesis [47]. Differential expression of seed proteins between drought tolerant and susceptible genotypes following water stress was observed in peanut leaf proteins, which play a major role in photosynthesis [48]. Proteomics has been successfully used to understand the metabolic processes that occur during seed filling. Soybean seed proteins were analyzed at five developmental stages by using 2-DE and a semi-continuous MudPIT coupled with LC-MS analysis [49].
Biotic factors: A comparative proteomic analysis was carried out in two lines of Lycopersicon hirsutum harboring two different quantitative trait loci (QTL) that control resistance to bacterial canker, suggesting that these two QTLs may confer resistance to bacterial infection through distinct mechanisms [50]. A comparison of the protein profiles of the root hairs and roots showed 96 differentially expressed proteins, of which only twelve were unique to root hairs. Moreover, this study showed that soybean root hairs could be a good starting material for further proteomic analysis in the study of symbiotic interaction with Bradyrhizobium japonicum [51].
Several studies suggested the application of proteomics in detecting unintended effects in genetically significant modified crops and changes in gene expression profiles of transgenic crops [52,53]. Transgenic integration may result in deletions, insertions, and rearrangements, which in turn may influence harmful effects [54]. These can be predicted by targeted analysis of transgene integration sites and related metabolites; however, they do not necessarily predict changes in food composition and quality; therefore transcriptomics profiling is limited in evaluating unintended effects.
Proteomic technologies have also been applied to investigate genesilencing products in transgenic research. Substantial suppression of GlymBd 30 K, a dominant allergen of soybean seed, by a reversegenetic approach has been confirmed by proteomic analysis [35]. No significant changes were observed in the polypeptide pattern of the transgenic seeds compared with the proteome map of the nontransgenic seeds. This suggests that transgenic-induced gene-silencing technique successfully removed the allergen from soybean. Several other allergen proteins include profiling in olive pollen, which showed polymorphism associated with physiochemical differences in response to different stress or physiological conditions [55,56].
With the commercialization of GM crops, these unintended effects are one of the most controversial issues on the biological safety of GM crops. The increasing use of “omics” technologies significantly contribute to our understanding of the biological safety of transgenics, since a systematic molecular analysis of GM crops is needed to address the unintended genetic effects [57,58].
In the process of protein biosynthesis, post-translational modifications (PTMs) (i.e. phosphorylation, glycosylation, sulfation, prenylation, acetylation and ubiquitination) are significant in altering protein functions, which include stability and localization, by binding to amino acids [59]. Experimenting with PTMs showed that the functions of a protein are well conserved; however, the PTM positions may not be stationary over evolutionary time.
PTMs are not stable under normal conditions for proteomics or MS, thus making them difficult to study. However, they regulate many cellular functions, such as protein activity and signaling throughout the cell. PTMs are identified by purification or protein separation using polyacrylamide gel electrophoresis (PAGE). The function of PTMs is currently being studied by analyzing the cell response to stress.
Phosphorylation
Reversible protein phosphorylation affects metabolism, the cell cycle, stress, hormonal responses, stomatal closure, development, as well as cytokinesis in the plant cell [60]. Currently, plant phosphoproteome studies are carried out through Immobilized Metal Chromatography- IMAC [61], precipitation [62], metal oxides, such as TiO2 [63] and ZrO2 [64]. Some proteins may be found in more than one organelle while others may be targeted to membranes via lipid anchors [22,65]. The study of subcellular fractionation makes it easier to understand specific phosphorylation events because the site can be determined. Phosphorylation has been proved to be involved in the cells response to fungal elicitors.
Ubiquitylation
Ubiquitylation is responsible for the tagging of proteins that need to be degraded. It is a process where the C-terminal carboxyl group of an Ub polypeptide is conjugated to the target protein via an ε-amino group of one or several specific lysine residues within the target protein [22]. It requires an ATP-dependent Ub activating enzyme (E1), an Ubconjugation enzyme (E2), and an Ub ligase (E2), which recognizes substrates.
SUMOylation
SUMO (Small Ubiquitin-related Modifier) is as effective as a polySUMO chain and is involved in transcriptional regulation, DNA repair, nuclear transport, mitochondrial fission, and regulation of receptors at the plasma membrane [66]. The enzymatic process involved in SUMOylation is similar to that used in ubiquitylation except “SUMO needs to be proteolytically processed by the Sentrin-specific Protease (SENPs) to expose the C-terminal GG before it can be activated” [22]. In fact, in yeast and mammalian cell studies, there is increasing evidence that ubiquitylation and SUMOylation pathways interact with each other [67].
PolySUMO chains are formed from SUMO (small ubiquitin-like modifier) chains by proteins containing SUMO interaction motif (SIM), i.e. the SUMO consensus modification motif (ΨKxE/D) [68,69], in response to processes, such as DNA damage or meiosis [70]. Studies by Bruderer et al. suggest that until now, more than 300 putative polySUMO conjugates from cultured eukaryotic cells have been identified. Furthermore, both polySUMO and SUMOylation modify proteins at specific stoichiometries [71].
Glycosylation
Glycosylation can be N-linked or O-linked, which is determined by the transfer of glycosidase and glycosyl between the golgi apparatus and the endoplasmic reticulum. N-linked glycosylation is found abundantly in mammalian systems and is characterized by N-acetylglucosamine (GlcNAc) linked to Asn by an amide. However, detecting glycosylation is challenging because of the abundance of proteins, the wide range of diverse structure, fragmentation during collision, and large size, which sometimes exceeds what can be detected by the mass spectrometer.
Several techniques have been adapted to enrich glycoprotein/ glycopeptides [72] such as hydrophilic interaction liquid chromatography (HILIC), which detects glycopeptides by using TiO2 and uses the hydrophilic nature of glycopeptides [73]. The experiments based on the detection of glycosylation in plants have mainly relied on 2D-PAGE to separate affinity-purified proteins or to separate total protein allowed identification of 102 proteins, of which, 94% were predicted to be secreted proteins [74]. Glycosylphosphatidylinoitol (GPI) anchored proteins are essential for cell viability and stable localization of the protein to a biological membrane. GPI consists of a phosphoethanolamin linker, a glycan core, and a phospholipid tail (the functional groups), phosphoinositol, glucosamine, and mannose (the core). GPI anchors are difficult to identify, however, the few studies that have been carried out solubilized isolated membranes with the detergent triton X-114 while alternating temperatures [75-77].
Redox-mediated modifications
Redox mediated modifications involve S-nitrosylation, Tyrosine Nitration, and S-glutathionylation. Nitric oxide (NO) is utilized in the regulation of neurotransmission, and immunological and inflammatory responses, in both animals and plants [78]. It is mainly produced by nitric oxide synthase [22] and reacts rapidly with O2-, or glutathione (GSH), generating ONOO- and S-nitrosylated glutathione (GSNO). Because of the three ways of nitration, the proteins modified by such processes are abundant. However, s-nitrosylation is in low abundance and reacts rapidly with various chemicals, therefore, it is difficult to detect.
In plants, nitric oxide affects germination, leaf expansion, lateral root development, flowering, stomatal closure, cell death (hypersensitive response), and defense against biotic and abiotic stresses [79]. Additionally, S-glutathionylation affects flowering, differentiation, cell death, symbiosis, and pathogen resistance. During oxidative and nitrosative stresses, glutathione can modify redox sensitive cysteines and function as protection against irreversible oxidation [80].
Redox proteomics defines the PTMs of proteins by abundant reactive oxygen species (ROS). The zone of redox proteomics is a difficult area for comprehensive analysis because of the varied amount of ROS that create the modifications as well as the extent of independent amino acids along the protein sequence. Changes in peptide mass that show variance in the expected peptide arrangement of the original amino acid configuration is used to show the characteristics of the alteration. For example, Sheehan et al. [81] has used conversion to SO3H by adding 32 Da to the peptide mass.
The oxidation of mitochondrial proteome in the rice leaf has been studied by researching the carboxylation protein configuration of proteins in the organelle succeeding in vitro oxidation. DNP (an inhibitor) was administered to specifically identify the oxidized proteins and proceeding MS and 2D-PAGE parting, which was followed by a mass spectrometric study used to examine the oxidized proteins. Our study confirmed that oxidation damage of the proteome is linked to peach fruit decay via mitochondrial function [82].
Post-translational modifications
Protein alteration through the accumulation of a lipid moiety to the protein has been testified extensively in organisms. Every kind possesses its own precise biosynthesis enzymes for the creation of the post-translationally adapted protein. Numerous pharmaceutical agents that inhibit the prenylation of these proteins have been advanced to oncology treatments. The lipid-modification of GTP gases in plants is viewed and connected to cellular growth and indication through their effect on stomatal closing brought by abscisic acid [83].
The organelle constituents are an important determining factor when studying the structure and function of a cell. It is necessary to demonstrate the specific localization of a protein for an organelle protein catalogue to be useful in terms of cell biology studies. Organelle isolation was initially used to associate proteins with their respective organelle through the differential centrifugation and density gradient fractionation methods. Through these methods many protein components of numerous organelles, such as the lipid raft, endoplasmic reticulum, mitochondria, plasma membrane, and nuclear envelope have been identified [84-86]. The success of these methods, however, depended greatly on the purity of an organelle, which was difficult, if not impossible, to obtain from subcellular fractionation approaches. Organelle proteomics is relevant because the functionality of proteins and cellular mechanisms are clearly linked to their subcellular location. Protein data available from various databases including AMIGO and SWISSPROT was used to build a subcellular location database for Arabidopsis proteins [87]. The database comprises of ten distinct subcellular locations and over six thousand proteins. Currently, several datasets of organelle proteomes such as Arabidopsis mitochondrial database (AMDB), Genome organelle database (GOBASE), and Plant organelle database (PODB), are developed to resolve protein association with multiple organelles and identify changes and protein-organelle association during the study [88-90].
The use of affinity chromatography following the process of subtractive proteomics to purify the organelle greatly enriched targeting organelle-specific proteins [91]. This advantage has led to a new method of accurately profiling protein organelles. Protein distribution among sub-cellular fractions can be monitored with quantitative proteomics. These distribution patterns can then be used to assign proteins with similar patterns to the same organelle [92].
Chloroplast and mitochondrial proteome
Chloroplasts perform basic functions including photosynthesis and amino acid biosynthesis. Most of the proteins of chloroplast are known to encode in the nuclear genome and imported into chloroplast upon translation. Therefore, understanding the chloroplast proteome will be of high value to predicting pathways to define regulatory levels of gene expression. Using tandem MS about 690 proteins were identified from Arabidopsis chloroplasts [93]. The chloroplast proteomes contains many proteins that are of unknown functions and not predicted to localize to the chloroplast. Further studies on transcript profiles of the chloroplastencoded proteins would be required to determine the expression of nuclear encoded chloroplast genes and their regulation in various pathways. Mitochondrial sequences in plants are notably different from those in other model organisms, such as yeast (Saccharomyces cerevisiae), due to additional components, non-phosphorylating bypasses of the electron transport chain, and specialized metabolite carriers, making analysis by bioinformatics tools challenging [94]. Since the need arose, Arabidopsis has been analyzed using TargetP predicting up to 2,897 nuclear-encoded mitochondrial-targeted proteins [4]. Millar et al. [95] analyzed the Arabidopsis mitochondrion by 2-D gel separation, identifying 100 abundant proteins and 250 low-abundance proteins. Later, using LC-MS method, Salvato et al. [96] identified 1060 proteins in Solanum tuberosum mitochondria.
Nuclear proteome
The eukaryotic nucleus serves as the regulatory hub of the cell and a repository of various macromolecules. Apart from DNA replication and transcription, the nucleus is important for cellular homeostasis and the determination of the genomic response to stress tolerance [97]. Given the central role of the nucleus, a detailed proteomic analysis is necessary to better understand its protein content, intracellular distributions, concentrations, turnover dynamics, the protein–protein interactions, and PTMs responsible for their function. An attempt at the complete coverage of the plant nucleus was made in Arabidopsis using 2-DE and MALDI-TOF MS analysis [98]. Their study identified 158 proteins with various cellular functions as well as 54 proteins that were either differentially up or down-regulated based on spot intensity in response to cold stress, thus supporting the regulatory role of the nucleus.
Cell wall proteome
Plant cell walls play an important role in defense and development. About 400 cell wall proteins (CWP) were identified in Arabidopsis [99]. The isolated CWPs in rice were evaluated for contamination by cytosolic proteins by measuring the enzymatic activity of an intracellular marker, glucose-6-phosphate dehydrogenase [100]. Furthermore, comparative analysis with known Arabidopsis revealed 25 novel, rice-specific CWPs. Different methods were tested for the extraction of proteins from the cell wall enriched fraction (CWEf) obtained from a sample formed by skin and seeds of ripe berries of Vitis vinifera [101]. The comparison of 2-DE reference maps of protein extracts from CWEf and CYf indicated the presence of both common and unique traits. Forty-seven spots were identified in Vitis berry, of which some were found to be cell wall proteins, while others were proteins not traditionally considered as localized in the apoplastic space. Future work should include: extracting and identifying CWPs still recalcitrant to proteomics, describing the cell wall interactome, improving quantification, and unraveling the roles of each of the CWPs.
Secreted proteome
The secretory proteins assist in many of the key functions of the cell, particularly for bacteria. Proteomics has helped reveal approximately 90 extracellular proteins in the gram-positive bacterium, Bacillus subtilis via genome-based models. This analysis led to the discovery of the cytoplasmic cell envelope proteins as well as confirming the significance of the secreted proteome in the formation of biofilms, which will assist in our understanding of the virulence of pathogens such as B. subtilis [102]. Furthermore, the use of proteomics has helped unveil proteins in the cell wall of Arabidopsis that typically reside in other organelles as well as previously unknown extracellular phosphorylation in plants. This study was completed using CaCl2 for fractionation, separation by 2DE, and identification by genomic database searches along with MALDI-TOF MS [103].
Endoplasmic reticulum
The endoplasmic reticulum (ER) is a specialized endomembrane system that is central to a number of biological functions such as protein folding, sorting, secretion, intracellular calcium regulation, protein N-glycosylation, and the storage of proteins and lipids [104]. Maltman et al. [105] were the first to investigate proteomics of plant ER by comparing the complexity and differences in the proteome between germinating and developing castor bean endosperm. Similar to mammalian and yeast systems, plants exhibit retrograde and anterograde transport by mechanisms mediated by Coat Protein complexes (COP I&II) at specialized areas known as ER export sites. While soluble proteins are exported from the ER by a bulk flow mechanism, the export of membrane-spanning proteins from the ER and subsequent sorting to other locations appear to be more complex [106]. Comparative studies have shown that the plant secretory pathway is by no means identical to the pathway in yeast or mammals and that many plant-specific features remain to be explored. Despite the central role of the ER and Golgi apparatus in cell metabolism and secretory pathways, very few proteomic studies have been reported on these endomembrane systems due to difficulties associated with their isolation and purification.
Protein–protein interactions form the basis of a large number of cellular processes. Genetic suppression has proven to be a powerful tool in identifying functional interactions. With advances in proteomics, more studies are now being directed towards unraveling the subtle network of interactions that govern cellular processes and development in organisms [107]. However, the many bacterial genomes that have been sequenced in the recent past have not left us with clues regarding the functions of more than half of their genes. Though a number of biochemical approaches have been successfully employed in determining protein–protein interactions in vitro, they essentially involve perturbation of the protein by insertion of a probe [108]. Genetic suppression obviates the need to modify the protein and provides alternative means to unveil the functional relevance of interactions in vivo.
In the cell, proteins are defined by their location and timing to carry out activities such as protein degradation in proteome complexes. These complexes have been analyzed using a multitude of different methods in several model organisms to map protein-protein interaction [109]. For example, yeast, Drosophila, and human protein complexes have been studied by large-scale affinity purification followed by mass spectrometry, and human genome-wide interaction maps using Yeast-2-Hybrid (Y2H) screening [72,110]. This study displayed binary interaction information, indicating that these proteins were tested in pairs [111]. Furthermore, this mapping displayed many transient complexes essential to extracellular signaling and correction of misfolded proteins [112]. It is well known that protein-protein interactions play a fundamental role in internal equilibrium maintenance [113].
Protein-protein interaction (PPI) studies for differentially expressed leaf proteins to water stress have been done in peanut using Domain Interaction Map (DIMA), which finds functional and physical interactions among conserved protein-domains. All the accessions were queried using the list of protein family (PFAM) identifiers. The integration of evidence from different sources involves analyses using the domain phylogenetic profiling and domain-pair exclusion method for predicting domain interactions from experimentally demonstrated protein-protein interactions using IntAct [114] STRING [115] and domain contacts from crystal structures using iPFAM [116]. Phylogenetic profiling methods revealed that many of these proteins have potentially predicted functional partners. The Domain Pair Exclusion Method (DPEA) was used to derive the most likely domaindomain interactions from experimentally supported protein-protein interactions using IntAct. Recent integration of genomics assists in the study of protein-protein interaction by predicting or confirming the aforementioned.
Protein-Protein interaction analysis
Mathematical gene interaction network optimization software (MINOS) was used to estimate protein interactions in the tolerant and susceptible peanut cultivars (unpublished data) based on the S-system differential equation that simulated an expression profile [117].
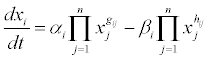
Where: xi: ith protein expression; i: ith protein production velocity coefficient; gij: coefficient of ith protein production velocity and jth protein expression; i: ith protein degradation velocity coefficient; hij: coefficient of ith protein degradation velocity and jth protein expression.
Protein orthologs were further identified in Arabidopsis and those interacting with them were analysed using Arabidopsis thaliana protein interaction database [118].
Metabolite profiling provides direct functional information on metabolic phenotypes and indirect functional information on a range of phenotypes that are determined by small molecules. A key aspect of metabolite profiling is that it can be used in high-throughput operation and provides a valuable combination of high performance and low unit cost per sample.
Metabolite profiling has now been accomplished in various plants, including Arabidopsis, Medicago etc., [119-121]. In plants, numerous genes have been interpreted on the basis of relationships between transcript and metabolite levels [122].
Metabolite profiling as an integral component of genomics
Integrated genomics has recently included metabolite-based approaches [123]. Plants expressing only parts of metabolite and transcriptome of transgenic versus wild-type plants showed pleiotropic perturbation of global transcript abundance, precursor accumulation, disturbed protection from UV rays, and a morphological phenotype. The expression of the complete pathway resulted in attenuated changes in growth behavior, metabolite and transcriptome composition [124]. Integrated approaches are also useful in the systematic characterization of biological processes.
Metabolite profiling as a diagnostic aid
Metabolite profiling is widely used as a diagnostic tool to determine the mode of action of various herbicides based on Gas Chromatography (GC) coupled with MS [125]. The analyses use the statistical tools of hierarchical clusters and a principal component analysis to compare large data sets. Studies suggest that the approach of headspace GCMS with statistical discriminant analysis could prove to be useful in identifying the compound class to which unknown MSTs belong [126].
Bioinformatics is concerned with the acquisition and storage of analyses and genome based information. There is a strong inclination to implement such pipelines using mechanisms developed as open source software, which facilitates the reuse and sharing of expertise. A diversity of approaches has been applied to explain eukaryotic genomes. They use comparative genomics, which relies on the ability to deduce organizational, structural or functional biological significance. This information is derived from sequence data arising from one or more taxa. Computational tools are mostly used as central aspects of comparative genomics, as it is in silico discipline. One of the methods for detection of homologous regions has been used to the complete Arabidopsis genome and concatenated rice. Most of these studies, such as Genome Pixeliser, has successfully been used to visualizing relationships and analyzing the evolution of encoding resistance genes in Arabidopsis among other genome duplication events.
Bioinformatics also uses the methods of data mining, the process of discovering knowledge in databases. Recently, the majority of data mining work in bioinformatics is founded on the requirement to screen through large, usually sequence-based, data sets looking for homology. The foundation of such data sets is sensitive to the exact and functional organization of any original explanation. Software tools are now being made available to aid the biologist in navigating existing ontologies, and in contributing their own data in the context of the wider group efforts [127,128].
The focal point for plant proteomics has, to date, been in development of species-specific databases, such as the Rice Proteome database containing indication maps based on 2DE-PAGE proteins from rice tissues and subcellular sections [129]. The keys to integration of metabolomics are bioinformatics tools and methods of analysis using biochemistry, plant physiology and ecology. This is mainly evident in the need to visualize and replace data, and develop an understanding of metabolic routes.
The multiple-omics based approach involves building a database schema that integrally stores and manages the multiple-layered biological information. Figure 1 shows a concept of the biological and hierarchical data integration. In the schema, data will be collected on biological multiple hierarchies and located on the directed graph of each hierarchy. Consequently, a biological mechanism, such as abiotic stress response, will be inferred as a network system across multipleomes.

Figure 1: Concept of the biological and hierarchical data integration.
The data of transcriptome and metabolome was also stored in the database, and the relationship between the data in different biological hierarchies was determined. Some metabolic systems were found, which include up-regulating metabolites under flooding stress based on the temporal profile analyses. Furthermore, tag information that concisely showed the feature of the profile (up-regulation, downregulation and/or existence of the maximum or minimum during the observed time period) was added one by one to the temporal profile data in the database. The user of the database can easily retrieve the data based on the feature of the time profiles. An omics table integrated in the database revealed time-variant mRNAs, proteins, and metabolites under flooding stress; and relationships across multiple OMICS, such as a protein product from an mRNA, and an mRNA or protein relating to a substrate or enzyme in a reaction including the metabolite.
With recent advances in the technology, currently, more genomic resources are available to improve breeding strategies and enhance crop productivity. Furthermore, advances in proteomics and bioinformatics tools have increased our understanding of the function and metabolic pathways of the molecules. In the past few years, there has been significant progress in plant proteomics studies due to the advances in protein isolation, separation methods and high-resolution using software tools and bioinformatics. This information is currently not limited to model plants, but even to plants known to be recalcitrant. This paradigm switch enhanced our ability to identify low abundant proteins and identify sub cellular localization, PTM, interaction and dynamics of changes relating to metabolite profiles. Systems biology provides the mining knowledge to integrate multiple OMICS data and test the expression of proteins as well as metabolites. As the sophistication of technology increases, several efforts have been made to create bioinformatics software tools and repositories such as Blast2GO, MINT, BioGrid, GelMap, and IntAct. We use the study of proteomics to further our knowledge of gene function on a genomewide level. It depends on appropriate model systems to analyze the extremes of certain structures in order to view the same structure in other systems in smaller amounts. Additionally, these databases give us a more intimate insight in the study of proteins involved in interactions that cannot be determined from the genome alone, such as PTMs. Now the focus will be on integration of transcriptome, proteome and metabolome data sets to integrate into large scale systems biology to unravel to signal transduction pathways at transmembrane level.
The author Keyura Katam acknowledges NSF for providing summer research opportunity and financial support.