Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Review Article - (2013) Volume 2, Issue 1
Keywords: Mi-2/NuRD, mTOR, Cell metabolism, Ageing, Cancer reprogramming
Ageing and cancer share some common biology [1]. Genomewide examinations show that overlapping genes between them can be classified into functional categories and pathways, e.g., cell cycle regulation, metabolic processes, DNA damage response, apoptosis, the P53 signalling and chemokine signalling pathway, and the immune/inflammatory response [2]. Currently, both the cause-effects “mutation” theory of cancer and the free radical theory of ageing are under question [3-5]. The former has difficulty explaining how cancers arise, and why conventional chemotherapy succeeds or fails [6]. Similarly, the idea that the macromolecular damage from toxic reactive oxygen species is the cause of ageing now faces challenges from evidences regarding the rival mTOR-centric model [3,7,8]. Oxidative stress has some signatures in cardiovascular ageing [9], but clinical trials with various combinations of antioxidants have failed to obtain the expected results [10]. Thus, novel concepts regarding the mechanisms of ageing and cancer are welcome [11]. Cancer was proposed to be viewed as a programmed, evolutionarily conserved lifeform, encouraging different thinking around its treatment [12].
As other experts previously proposed, I consider, that carcinogenesis is the speciation based on ageing host with maternal effects and beyond the host in figure 1. Breaking the rule of “Don’t put all eggs in one basket”, particularly the “sharing” interaction and diseased multicellularity, may cause ageing and cancer; the reprogramming of ageing and cancer is feasible via modulation of master regulators in ageing and cancer, such as Mi-2/NuRD, mTOR kinase and cell metabolism.
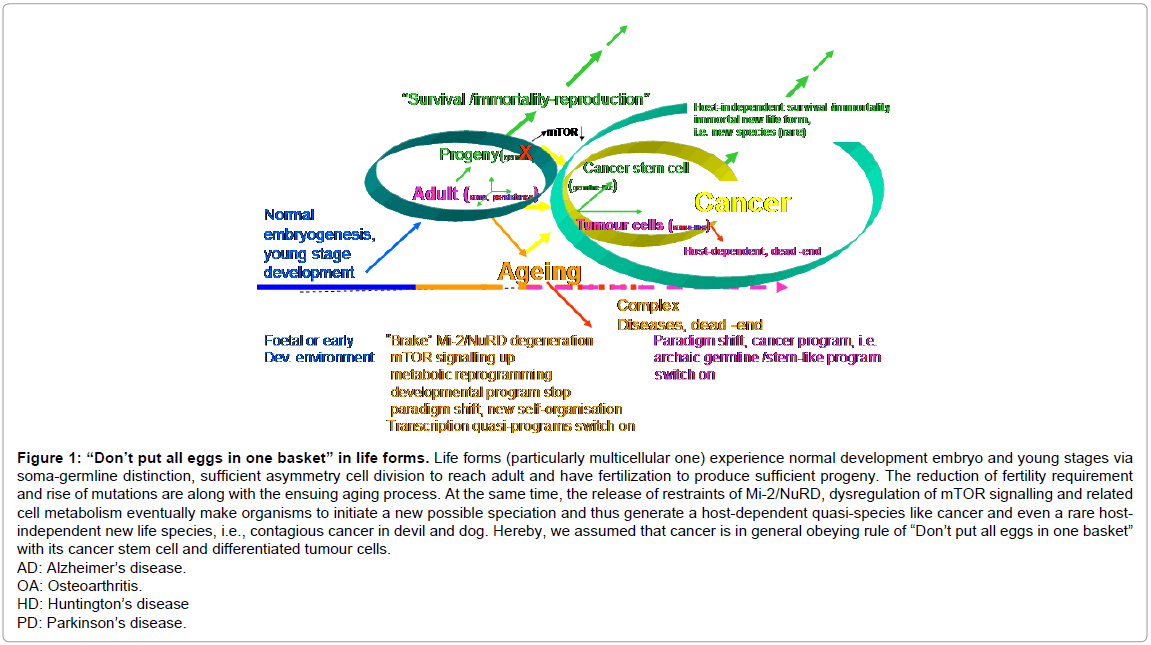
Figure 1: “Don’t put all eggs in one basket” in life forms. Life forms (particularly multicellular one) experience normal development embryo and young stages via soma-germline distinction, sufficient asymmetry cell division to reach adult and have fertilization to produce sufficient progeny. The reduction of fertility requirement and rise of mutations are along with the ensuing aging process. At the same time, the release of restraints of Mi-2/NuRD, dysregulation of mTOR signalling and related cell metabolism eventually make organisms to initiate a new possible speciation and thus generate a host-dependent quasi-species like cancer and even a rare hostindependent new life species, i.e., contagious cancer in devil and dog. Hereby, we assumed that cancer is in general obeying rule of “Don’t put all eggs in one basket” with its cancer stem cell and differentiated tumour cells.
AD: Alzheimer’s disease.
OA: Osteoarthritis.
HD: Huntington’s disease
PD: Parkinson’s disease.
Are ageing and cancer inevitable and/or reversible?
Mature cells can be reprogrammed to become pluripotent–the discovery for which Dr. Yamanaka and Dr. Gurdon won the 2012 Nobel Prize in medicine. The Waddington’s epigenetic landscape hypothesis has gained popularity in induced Pluripotent Stem Cells (iPSc) [13,14] and the Self-organisation Map (SOM) program embedded cell attractors studies in cancer research, for example tumor heterogeneity [15,16]. Reprogramming could be significant for our understanding carcinogenesis and ageing, and it theoretically suggests that reversals of ageing/cancer might be possible.
Weissmann’s germ-soma distinction hypothesis [17] includes the ageing-fertility trade-off concept [18-20] along with the proposal of “Nothing makes sense in biology except in the light of evolution” [21]. We postulate that life systems are spatiotemporally struggling for survival and broadly-defined immortality. Spatially, systems may try to occupy as much room as possible by producing more progeny and more cell growth and proliferation; temporally, they strive for as long a lifespan as possible. “Don’t put all eggs in one basket” means diversifying the options. While pursuing the long-term survival or immortality, the gonad or germline cells can emit the signal, an organism could thus need sacrifice its cell growth, cell proliferation or lifespan, this eventually makes ageing and cancer inevitable [22,23].
Two cell attractor theories have been recently updated: the GRNSOM cell attractors theory [24,25] with an emphasis on the high dimensional nonlinear systems and of self-organized behavior; on the contrast, the “metabolism-first” Independent Attractors (IA) theory [26,27] which emphasizes interactions between active gene products (mainly proteins) and independence of the genomic sequence are descended from a pre-cellular state from which life originated. As IA model is based on the cell as an open system, the attractor acts as the interface between the cell and environment. Environmental stressors can serve to trigger attractor and therefore phenotypic transitions. Based on these, and taking into account various levels of self-organization, such as individuals, population, we have described the CRS-related GRN-SOM cell attractor hypothesis to understand diseased multicellularity-induced carcinogenesis [28-31]. Clinically, epigenetic reprogramming may reverse some cancers through targeting the components of Mi-2/NuRD with histone deacetylase (HDAC) 1 or Dnmt1/2 inhibitors [32].
~3.8 billion years ago (BYA), there would come earliest chemical evidence of life. Many principles of chemistry and physics including self organisation system (SO) (i.e., complex systems are capable of adapting themselves completely at each instant to the results of their analysis of external conditions and previous performance) can be applied on life systems, which evolved from chemical and physical world. I argue that, similarly, cancer comes from its host, owning many traits of host, but likely as intermediate or new specie since we may consider cancer as a “jump from the sinking ship (host)” as a different life form [33,34] (Figure 1). The ageing of the host is here surmised to provide the resources for and the possibility of carcinogenesis [35] and thus speciation. If it could be the case, we can definitely find out the different systems governing the host and cancer and targeting the cancer and leave the host healthy [36]. Besides, the cancer “avalanche” can be triggered by a minor event with little significance of its own. However, due to ageing and environmental stresses, at one time point, a few cells reside in a sub-critical state and ripe for collapse and restructuring, so paradigm shift. After the catastrophe, the system (somatic cells) moves itself into a cancer “stem-like” states, such new sub-critical state with proliferation descends to cancerous state (Figure 1). The sequential multi-stable states happen essentially among a random process, and thus exist on the edge of chaos. The studies of self-organized oscillations and chaos have already provided some novel insights [37-39]. This notion is compatible with the GRN-SOM cancer attractors theory [24,40] and the CRS-linked GRN-SOM cell attractor hypothesis [31,41]. Alongside ageing/cancer, the whole life system could be arbitrarily defined as “survival “with its progeny’s continuity (Figure 1). However, as a life system is one dissipative system [27,42], an interruption in external energy ( i.e. the price) could logically delay system failures during ageing/cancer processes. According to Huang, “Nothing in evolution makes sense except in the light of (systems) biology,” implying the whole system rather than single gene is being critical for complex diseases [43]. Through systematic epigenetic modulation of the genome GRN, human cell and tissue reprogramming engineering could deal with the inevitability of ageing/cancer [44].
The origin of modern life and the rise of mitochondria
There are many different angles from which to look into the basis for reversing ageing/cancer. Although the details much remain to explore, Mi-2/NRD likely orchestrates asymmetry cell division, “sharing” interaction, chaotic and oscillatory gene expressions, sex, stemness, tumor heterogeneity, ageing damage heterogeneity [45-47]. The factors, which are under the regulation of Mi-2/NuRD or other CRS, ensure the robustness of multi-cellularity among the cell communities [41].
Alongside epigenesis, such as Mi-2/NuRD linked-processes, the mitochondria also contribute to ageing/cancer. Many factors associated with mitochondria are longevity/ageing genes, particularly mTOR (or TOR) [11,48,49]. Since the IA has a “metabolism-first” notion embedded [26] and emphasizes the significance of environmental cues and proteins, we extend our CRS-GRN cell attractor concept to the interplay of Mi-2/NuRD with mTOR and cell metabolism in that mTOR is an environmental sensor, and a master regulator for the translation process and also for recycling and refolding the protein aggregates through autophagy. Besides, the metabolism itself can probably get involved the chemical network, information flow and interaction. The cause-effects “mutation theory” says nothing about the metabolic derangements common to virtually all cancers [6]. Here, we consider a focus on their “sharing” interaction (rather than keeping all in their own hands) with fittest comparative advantages during major evolutionary transitions.
First, the origin of modern life might have evolved via “sharing” [50] ~3 billion years ago (BYA). That is, a group of archaic life elements such as archaic cells, which individually differed in their comparative advantages and capacity for replication and metabolism [51] may have collaborated and fought to survive better than single units in the harsh environment. They may have cross-fed not only genetically but also metabolically without competition-possibly a global mega-organism [52]. It is not individuals but the community of those that “share” that survives and evolves. It was the communal ancestor of modern life [50]. Coincided with the appearance of oxygen in the atmosphere, around 2.9 BYA [52], the mega-organism has broken apart as three. In modern cells, the fine system need for such functional “sharing” network, possibly including Mi-2/NuRD [31]. Interestingly, the MEP- 1, an interactor of Mi-2/NuRD, controls gene expression via the 3’ untranslated region (3 UTR) in the worm Caenorhabditis elegans [53], which may be similar to archaic microRNA repression [54] in predated RNA system in “proto-cells” in “RNA world” theory.
Another result of “sharing” is multi-cellularity, characterized by cell labor division and communication in an extra-cellular matrix ~0.9 BYA [27]. Some “altruistic” cells undergo programmed cell death to benefit the whole multi-cellular organism [31,55]. Further, mitochondria function as power stations for the cell, which is vital for evolutionary innovations and nucleus development. Endosymbiosis generated the mitochondria and restructured the distribution of DNA in relation to the bioenergetic membranes. This leads to a vast expansion in the number of genes expressed. This quantum leap in genomic capacity relied on mitochondrial power and paved the way to multi-cellular life [56]. Disruption of the “sharing” interaction, we could say, less chance keep the beneficial traits to cells, and in modern cells and organisms, loss of multicellularity may lead to cancer, and epigenetic therapeutic reprogramming needs to reverse this abnormality [31,57,58]. We stop here and keep from descending to further hypothesis. Loss of communication through the extra-cellular matrix or a link between LMNA and Mi-2/NuRD could cause premature ageing [59] and cancer [60,61]. While we treated cultured primary osteoarthritis chondrocyte cells with rapamycin, an inhibitor of mTOR signalling, cells dramatically reduced touching branches; so mTOR signalling could promote cell-cell communications and/ or cell growth alone (Zhang Y, unpublished observation). I speculate that the return to normal sharing connection may help the reversals of ageing and cancer. It is tempting to test whether a block of component of Mi-2/NuRD, mTOR signalling or metabolism in germline alone can affect the aging and to see what will happen with interruptions in somatic cells alone and/or in combination [62]. I am wondering if any surprise could be there. Keep in mind, some genes are not easy to manage since many mitochondria related genes have maternal effects and aging-related features, they will otherwise provide significant insights to ageing and cancer, which is out of scope of this article.
Mi-2/NuRD, mTOR and metabolism in ageing and cancer
Paradoxically, it is exciting to demonstrate that mutations in metabolic enzymes may be carcinogenic while the somatic “mutation” theory is under question. Mutations in several TriCarboxylic Acid (TCA) cycle enzymes are linked to human cancer: Succinate Dehydrogenase (SDH) is linked to hereditary paragangliomas [63], Fumarate Hydratase (FH) is linked to skin leiomyomas and renal cancer [64] and isocitrate dehydrogenase (IDH) 2 and IDH1 are linked to gliomas and acute myeloid leukemia [65-67], driving R-2-hydroxyglutarate synthesis [68]. The conditional knock-in mice for IDH1 mutations show more reprogrammed early hematopoietic progenitors [66,67]. Epigenetic programming via the Tet2 DNA methylase is involved in their activity [68]. The metabolic products of mutant SDH, FH and IDHs can inhibit the activity of a class of α-ketoglutarate-dependent enzymes and may lead to alterations in DNA methylation linked to the histone demethylases. Further, mutant SDH, FH, and IDH1 may induce pseudohypoxia [69,70]. MBD3/NuRD may regulate many target genes in embryonic stem cells, and MBD3 association with its in vivo targets requires 5-methylcytosine hydroxylase Tet1 and MBD3 to bind preferentially to hydroxymethylated DNA relative to methylated DNA. We may postulate that induced pseudohypoxia could reverse cells to “archaic” cell attractors, similar to single cell state [23,31,71,72].
Fuel signals from cell metabolism are crucial for the activities of the Mi-2/NuRDs in normal health, ageing and cancer. For instance, the energy of ATP is essential for remodeling the Mi-2/NuRD, and is generally provided through the glucose-pyruvate axis and the TCA cycle. Cell metabolism also ensures that cells have a reservoir for acetyl- CoA, which is important for balancing Histone Acetyl Transferase (HAT) and HDAC [73]. Moreover, the oncogenic activation of Myc promotes glutamine utilization. Furthermore, c-myc regulate glucose metabolism and stimulate the Warburg effect [74]. The LMNA1-linked NuRD may be a key modulator of these ageing-associated chromatin defects [75]. Other than LMNA1, in many contexts, Mi-2/NuRD will be transiently recruited by different TFs [46,76].
In C. elegans, fumaryl acetoacetate hydrolase, which catalyzes tyrosine metabolism and causes fumarate to cycle into the TCA cycle, is required for a normal lifespan [77] and has a role in protein aggregation in connection with heat-shock factor 1 (HSF1), the master regulator of the heat-shock response for environmental stress cues [78]. The enhanced HSF1 expression was associated with poor cancer prognosis in human through several processes, including metabolism and translation. HSP genes are integral to this program, e.g. uniquely regulated hsp70 via HSF1/NuRD [79]. Basically, HSF1 rewires the transcriptome in tumorigenesis. Furthermore, our assays have related HSF1 regulation of the promoter activity of one of the important ATP transporters, the multi-drug resistance gene 1 (mdr1) (Zhang Y, Supplementary Figure 1), expanding its activity to drug resistance. Moreover, over-expression of HSF1 can promote longevity in C. elegans [80]. Further, HSF1 is essential for protecting cells from the proteindamaging stress associated with misfolded proteins, partly through autophagy [81]. Activation of the deacetylase and longevity factor SIRT1 prolonged HSF1 binding to the heat shock promoter of Hsp70 by maintaining HSF1 in a deacetylated DNA-binding competent state [82]. Besides, mTOR may phosphotate HSF1 [83]. Lastly, those genes are also longevity genes, and those mitochondrial genes have maternal effects.
In normal cells, glutamine is used for the synthesis of amino acids and nitrogen for de novo nucleotide formation. Cancer cells, in contrast, secrete glutamine-derived nitrogen as waste [74]. The cellular uptake of L-glutamine activates mTOR. Certain tumor cell lines do not need it. L-glutamine flux regulates mTOR translation and autophagy [84]. Glutamine is a key factor that influences the axis of mTOR-linked autophagy. Autophagy generally promotes longevity in C. elegans [85] and mammalian cancer survival [86]. A network of 3M can be seen in figure 2.
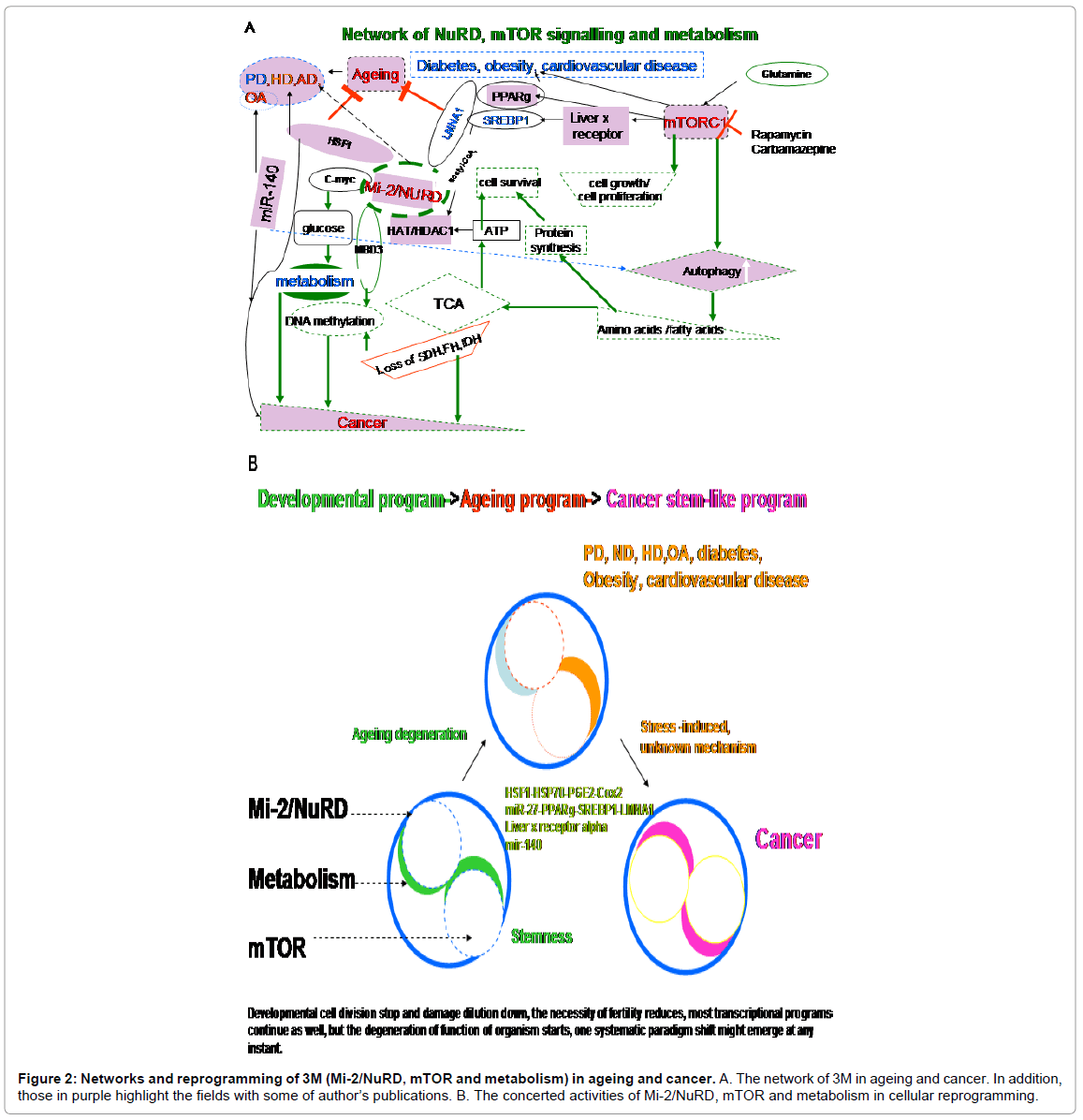
Figure 2: Networks and reprogramming of 3M (Mi-2/NuRD, mTOR and metabolism) in ageing and cancer. A. The network of 3M in ageing and cancer. In addition, those in purple highlight the fields with some of author’s publications. B. The concerted activities of Mi-2/NuRD, mTOR and metabolism in cellular reprogramming.
Concerted activities of Mi-2/NuRD and mTOR in reprogramming
Mi-2/NuRD can be recruited by many transcription factors for reprogramming [46,76] in different contexts. For example, Zfp281 mediates Nanog auto-repression through recruitment of the Mi-2/ NuRD and inhibits somatic cell reprogramming [87]. It is well known that HDAC inhibitors can significantly enhance the efficiency of cellular reprogramming [88]. Vitamin C has been shown to enhance reprogramming significantly [89]. Additionally, treatment with an inhibitor of DNA methylation, such as 5-aza-2’-deoxycytidine, increases the efficiency of iPS reprogramming [90]. The major activities of mTOR include control of cell growth, cell proliferation and autophagy-related pathways. Dual targeting of mTORC1/C2 complexes enhances HDAC inhibitor-mediated anti-tumor efficacy in primary Hepato Cellular Carcinoma (HCC) in vitro and in vivo [91]. Some well-characterized mTOR inhibitors and autophagy activators (e.g. PP242, rapamycin and resveratrol) notably improve the speed and efficiency of reprogramming of iPSCs [92]. Cao et al. [93] have recently confirmed that the mTOR inhibitor rapamycin can efficiently prevent the accelerated geroconversion of human cells that is induced by progerin LMNA accumulation [94]. Similarly, [95] shows that mice deficient in LMNA had enhanced mTOR complex 1 signalling specifically in tissues linked to pathology. The reversal of elevated mTORC1 signalling by rapamycin enhances survival and improved the defective autophagic-mediated degradation in LMNA (-/-) mice. The use of rapamycin-like mTORC1 inhibitors (e.g., temsirolimus) activates autophagy and ameliorates cardiomyopathy caused by lamin A mutation [96].
Because mTOR signalling is up-regulated in HCCs, one recent study [97] showed that the two mTOR inhibitors (RAD001 and BEZ235) had synergistic effects on the inhibition of the proliferation of HCC cells. A large number of genes reverted to normal liver tissue expression in mice treated with both drugs. Also, the down-regulation of autophagy genes is seen in tumors in comparison with in the normal liver. On the other hand, LMNA mutations can affect the adipocyte differentiation pathway acting upstream to SREBP1 and PPARg [98,99]. In fact, cells harboring mutations in these genes accumulate an immature form of lamin A that can stably interact with a transcription factor SREBP1 involved in adipocyte differentiation [100]. Oltipraz regulation of the AMPK-S6K1 pathway also affects Liver X Receptor (LXR) activity and LXRα-induced lipogenesis, leading to the identification of AMPK and S6K1 as attractive targets for intervention of steatosis in the liver [101]. Activation of AMPK following oltipraz treatment resulted in decreased phosphorylation and inactivation of S6K1, a subsequent decrease in phosphorylation of LXRα by S6K1[101], decreased binding of ligand-bound LXRα to the LXRE upstream of sterol regulatory element-binding protein-1 (SREBP-1), and ultimate inactivation of SREBP-1 expression. DAF-12, one homolog of liver x receptor in the nematode C. elegans is one critical factor in ageing and regulates a connected network of genes to ensure robust developmental decisions [102]. The reduction in SREBP-1 expression contributes to reduced lipogenesis, fat accumulation, and hepatic steatosis [101]. The result is consistent with the previous observation that the induction of acetyl- CoA carboxylase (ACC) and fatty acid synthase (FAS) in the fatty liver patients is associated with SREBP-1 [103,104]. Hepatic fibrosis and/or carcinogenesis can be caused by a gain-of-toxic function with accumulation of the mutant Alpha1-Antitrypsin Z (ATZ) variant inside cells. The autophagy enhancing drug Carbamazepine (CBZ) decreased the hepatic load of ATZ and hepatic fibrosis in a mouse model of AT deficiency-associated liver disease [105].
Inhibition of Mi-2/NuRD for HDAC or knock-down of RBBPP48/46 facilitate direct conversion, for instance, the direct conversion of C. elegans germ cells into specific neuron types [106]. Broadly to say, the metabolism chemical network is also one kind of neuron network. Interestingly, LIN-1/ETS, an interactor of Mi-2/NuRD in C. elegans [107] has its role in Ras and Hox cancer-related signalling pathways. In mammals, three ETS TFs- reprogrammed amniotic fluid cells could treat vascular diseases. Finally, the histone demethylases Jhdm1a/1b are key effectors of somatic cell reprogramming downstream of vitamin C [89]. In general, we hypothesize that, alongside the dysregulated Mi-2/NuRD, mTOR signalling and cell metabolism, the sequential multi-stable states toward cancerous states happen essentially among a random process, the diverse reprogramming causes the tumour heterogeneity due to accidentally forced expression of TFs and thus those resulting reprogramming or the like.
Reversals of aging and cancer, and application of GRN-SOM cell attractors in cancer
Reversal cases of aging and cancer appear. Just name a few: for example, implanting young stem cells has rejuvenated ageing stem cells in the treatment of progeria, which is a disease that causes abnormally accelerated ageing and a deficiency of Lamina A [31,41,59,108]. The researchers injected stem-like or progenitor cells into progeriatric mice. Some transplant recipients had robust health and a lifespan up to 2-3-fold of the controls [109]. Another case using mice has shown that modifying telomerase activity can rejuvenate the tissue and organs [110]. Recently, a stem cell-based approach to ageing-related cartilage damage repair has been published [111].
Another significance of GRN-SOM attractor’s theory is to make prognosis. Gene Expression Dynamic Inspector (GEDI) patterns may be promisingly developed for identifying the onset and progression of cancer and ageing [24,25,41,48,112] (Figure 3). I made systematic analysis of cell-division mechanism with GEDI, strikingly, they have a consistently similar pattern when deficiency in either component in this mechanism (Zhang Y, in preparation). Therefore, I speculate, like weather forecasts, simple genome-wide expression patterns like GEDIs can be predictive, for example, predicting that the cancer “weather” will be “stormy”, “cloudy” or “sunny”. They are like impressionist paintings of a landscape, giving an overall image when viewed as a whole rather than capturing the fine details of paint chemicals. For sure, what is largely unknown to us is the genius from the painters and the beyond. However, further studies need to refine and identify the distinct patterns for different stages for clinical use. For some cancers, this might even involve running a PCR array with miRNAs and using the resulting data as a proxy for GRN, since the medusa-like pattern of GRN has been revealed in cancer subtypes, with particular miRNAs as dominators [113].
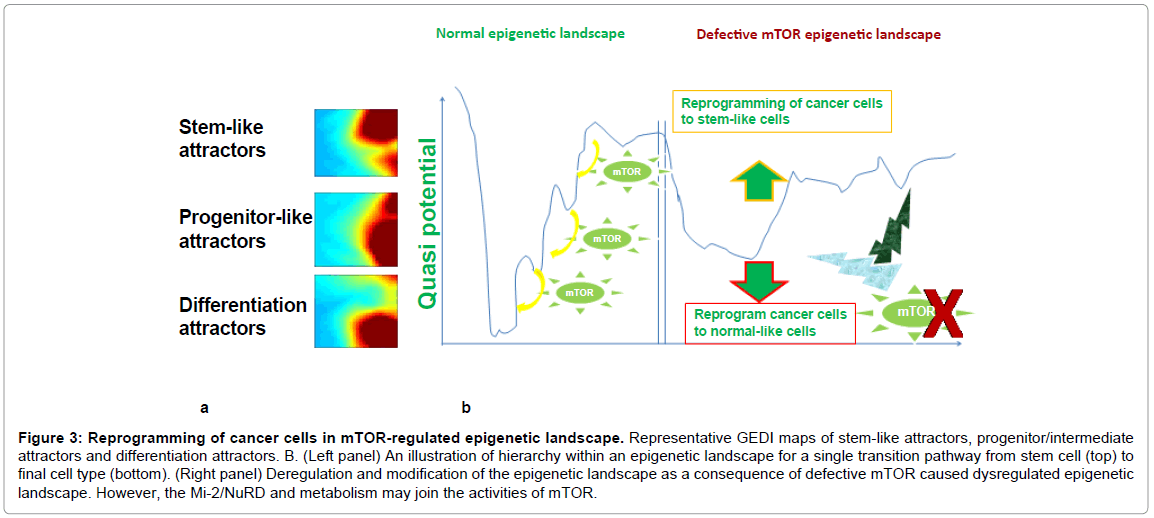
Figure 3: Reprogramming of cancer cells in mTOR-regulated epigenetic landscape. Representative GEDI maps of stem-like attractors, progenitor/intermediate attractors and differentiation attractors. B. (Left panel) An illustration of hierarchy within an epigenetic landscape for a single transition pathway from stem cell (top) to final cell type (bottom). (Right panel) Deregulation and modification of the epigenetic landscape as a consequence of defective mTOR caused dysregulated epigenetic landscape. However, the Mi-2/NuRD and metabolism may join the activities of mTOR.
Some gaps evidently exist, but these self-organisation insights in system biology point of ageing/cancer suggest that they both are inevitable and reversible or preventable. Breaking “Don’t put all eggs in one basket” and “self-organisation” promote ageing and cancer. Cancer itself may hijack the the system of host with or without switching their original functions. Inhibitors for Mi-2/NuRD and its functional partner CRS, mTOR could separately reprogram and reverse ageing/cancer. However, it is tempting to find out effective combination rapamycin/ mTOR inhibitors, vitamin C and HDAC inhibitors of low–dosage and less side-effect in such reversals. Simplified genome-wide expression patterns could be developed as ensemble markers for ageing- related diseases or cancer.