Journal of Clinical and Experimental Ophthalmology
Open Access
ISSN: 2155-9570
ISSN: 2155-9570
Research Article - (2016) Volume 7, Issue 5
Objective: Amplify the absorbance signal related to the lysozyme extracted from contact lenses as measured by high-performance liquid chromatography (HPLC); examine the purity of lysozyme and how much thermal denaturation affects its elution profile.
Methods: In trial chromatographic runs during which contact lens extracts had been injected into the system, the fractions collected between 4 and 5.5 minutes pointed to lysozyme as the eluting protein as indicated by a Western blot. Proteins were extracted from contact lenses in a 50:50 solution of 0.2% trifluoroacetic acid (TFA): acetonitrile (ACN). Each contact lens extract was separated into 2 aliquots. After vacuum evaporation, aliquots no 1 were dissolved into the initial mobile phase to produce an enrichment factor of 8. Aliquots no 2 were left untreated in the regular extraction solution. Calibration of the absorbance signal at 220 nm allowed measuring the lysozyme levels in chromatograms. Parallel injections of both aliquots into the HPLC allowed comparing their lysozyme content. Purity of the lysozyme extracted was evaluated by viewing its absorbance spectrum across peaks. Lysozyme solutions, previously heated to 80 and 100°C were injected and compared with control solutions. Differences in median lysozyme content between aliquots no 1 and 2 and in peak area of lysozyme heated compared to control were tested with non-parametric methods.
Results: Median lysozyme level measured in enriched and regular extracts differed significantly (39.8 and 21.5 μg, respectively). However, once corrected for different injection volume and concentrating factor, the mean enriched lysozyme level (21.4 μg) was close to the one found in the regular extract. Observation of spectral absorbance suggests that eluting lysozyme is free from contaminants. Median ratios of peak areas of heated lysozyme over peak area of control lysozyme differed significantly from 1 at the temperature of 100°C (0.91), but not at the one of 80°C (0.95) with the signed rank test. Even when heated at 80°C, the elution profile of lysozyme appeared less symmetrical compared to control and presented an additional inflection point. These subtle changes were increased at 100°C.
Conclusion: Enrichment of the lysozyme extracted from contact lenses and solubilized into the initial mobile phase improves the sensitivity of this chromatographic procedure. This chromatographic protocol coupled with UV absorbance spectroscopy can detect thermal denaturation at 80 and 100°C.
Keywords: Absorbance spectrum; Adsorption; Chromatography; Contact lenses; HPLC; Lysozyme; Spectroscopy; Tear protein
Tear film components adsorb more or less to contact lens surfaces depending on their mutual characteristics, their affinity with the material of the lens, the specific characteristics of subject’s tears and glands as well as with the regimen of care used if any. The adsorption of proteins [1,2], lipids [3], and mucins to contact lenses has been reviewed previously [4]. The tear film is a complex mixture of numerous proteins, some of which interact with the contact lens material. With its basic isoelectric point, lysozyme, a main tear film protein often used as a marker for protein deposition and lens usage [5], contains a predominance of positive charges at a physiological pH [6]. Therefore, it adsorbs to hydrogel lenses, especially those that contain negative charges due to their methacrylic acid content [7-11].
Early on, the adsorption on contact lenses of high levels of protein has been associated in patients with a higher risk of adverse reactions such as giant papillary conjunctivitis (GPC) [12]. Patients, asked to wear their lenses longer than manufacturer’s recommendations on one eye, presented more severe upper conjunctival papillae, upper lid conjunctival hyperemia, as well as limbal congestion compared to the compliant control eye. Extracts from their hydrogel lenses had higher protein content on the non-compliant side [13]. The accumulation of such deposits may contribute to induce adverse clinical effects, possibly contributing to discontinuation of contact lens wear. However, the process of protein adsorption on contact lenses is a very complex problem. Tear-derived pellicle, useful to enhance biocompatibility, should be distinguished from deposits that adhere to the lens surface or matrix and that are likely to cause problems [14]. Molecules at the surface are more likely to induce problems, but their quantitation is difficult because adsorption is a dynamic process. Lysozyme loosely bound to the surface of an ionic contact lens may desorb and re-enter solution [15].
Part of the lysozyme adsorbed to hydrogel lenses may be denatured and non-functional [16,17]. Active lysozyme may be assayed by measuring its functional ability to hydrolyze the bacterial glycosidic bonds of Micrococcus lysodeikticus. Therefore, a comparison between estimates obtained with this micrococcal assay and those obtained with other methods of total lysozyme quantification allows obtaining the active proportion [17]. Comfort is not correlated with the total amount of protein bound on hydrogel lenses across the four FDA groups [18]. However, in subjects wearing etafilcon lenses, there is a positive correlation between subjective comfort and percent of active lysozyme estimated in this lens [19]. Deposition of lipids occurs more than that of proteins on hydrophobic silicone hydrogel (SH) contact lenses, but the denatured fraction of protein could be proportionally more important with SH lenses [1,20].
The activity of the surface active lysozyme may be obtained by subtracting the amount of loosely bound lysozyme from the total active lysozyme in situ, as obtained with repeated assays of Micrococcus lysodeikticus. Surface active lysozyme molecules are more likely to induce a response from the wearer, because of the direct contact [15]. A comparison of this in situ method to a method based on the extraction of protein on commercial lenses revealed that methods based on extraction of proteins from contact lenses allow a quantification of lysozyme within the matrix of the polymer or in the underlying layers of lysozyme [21].
These studies indicate the importance of improving analytical methods of lysozyme detection in order to keep up with the development of materials or processes that resist the symptomatic adsorption of protein onto contact lenses or their denaturation, once adsorbed. In addition, modern contact lens wear is frequently associated with non-compliance of wearers who stretch their replacement schedule or overwear their lenses [22]. These behaviors may have an impact on comfort and vision, leading to dropping out of contact lens wear [23]. This is why improved methods to analyze modern contact lenses are needed.
Unfortunately, the effect of lysozyme denaturation on the outcome of most methods of lysozyme detection is not clear. A notable exception comes from the work of Hall et al, who showed the absence of any active lysozyme on contact lenses previously heated to 80°C, as opposed to unheated contact lens controls [15].
Keith et al. analyzed lysozyme samples by high-performance liquid chromatography (HPLC) [5]. The lysozyme signal of this method could be increased by concentrating the sample into the initial mobile phase prior to its injection into the HPLC system because of 1) increased concentration of the analyte and 2) reduced formation of complex interactions arising from differences in composition between the extract and the mobile phase [24-26]. This HPLC protocol could also be coupled with UV absorbance spectroscopy in order to evaluate the purity of eluting peaks [27], to detect compounds possibly co-eluting with lysozyme, or to confirm the identity of a protein by spectral matching with a known protein standard [28]. The spectral signature of the analyte is especially useful for identifying components close to detection limits [29].
The objectives of this project are: 1) to compare lysozyme levels measured in a contact lens extract to those of an extract from the same lens but concentrated into the initial mobile phase of the HPLC; 2) to analyze lysozyme peaks and evaluate peak purity using absorbance spectra, and 3) to investigate the effect of thermal denaturation of lysozyme on peak area, shape, and absorbance spectrum.
This study was approved by the internal Review Board for Health Sciences of the University of Montréal. All procedures adhered to the tenets of the Declaration of Helsinki. After explanation about the nature and the intent of the study, student clinicians of the University Clinic of the School of Optometry asked their patients disposing of their old soft contact lenses for the permission to keep their lenses for the study. In order to protect their anonymity, subjects consented by word of mouth to participate in the study. Then, students left the lenses in saline-containing separate test tubes, which were placed in an envelope. The envelope includes space to fill in the following information: the date, the sex of the contact lens wearer, the brand of contact lens, the manufacturer, and the approximate length of wear (in days) of the lenses.
Contact lens extracts
Whenever possible, lenses are presented in this paper using the USAN (United States Adopted Names {http://www.ama-assn.org/ ama/pub/physician-resources/medical-science/united-states-adoptednames- council/naming-guidelines/contact-lenses.page}) terminology for contact lens polymers. Lenses were kept at 4°C in saline (Sensitive Eyes Plus Saline Solution, Bausch & Lomb, Rochester, NY) before extraction. Lenses were then rinsed with a 0.9% saline solution and gently blotted on absorbent paper by an experimenter with gloved hands. Proteins were extracted by soaking each lens into 1.5 ml of a solution of 50:50 trifluoroacetic acid (TFA) 0.2%: acetonitrile (ACN) with continuous agitation in the dark during 16-20 hours [5]. After this incubation, lenses were removed from the solution and discarded whereas extracts were kept at 4°C until use. Contact lens extracts were not pooled together. Water distilled and deionized (Barnstead Diamond distiller and nanopure system, Dubuque, IA) was used in all experiments. All organic solvents as well as TFA, which were HPLC grade, were bought from American Chemicals Ltd (Montreal, Canada). All other chemicals, except otherwise mentioned were from Sigma Aldrich (St-Louis, MO). Lysozyme from chicken egg white was used for calibration, comparison of absorbance spectrum and thermal denaturation assays.
Enrichment of the extract
In a first set of experiments, extracts from each contact lens were separated into 2 aliquots. The first set of aliquots was evaporated by vacuum centrifugation (Vacufuge, Eppendorf, Westbury, NY). Remaining solids were solubilised into a smaller volume of a 0.1% TFA solution of 15% ACN: 85% water (initial mobile phase) in order to enrich its concentration by a factor of 8 times. The second aliquots were left unmodified in the regular extraction solution [5]. Both sets of aliquots were injected into a HPLC system. The concentrations of lysozyme in each aliquot were calculated from peak area (see analysis) in order to compare the chromatograms obtained with and without the preliminary concentrating step.
Instruments, injections and experimental procedure
Eluents were outgassed with a vacuum pump at least during 10 minutes prior to injections. Volumes of 50 and 10 μL of the regular and enriched extracts were injected into the column at room temperature using the partial loop fill and the μL pick-up modes of an autosampler. Samples lists included contact lens extracts, samples containing a known amount of lysozyme for calibration or verification, or various controls.
Samples were injected into a HPLC system (Agilent-Varian, Walnut Creek, CA) equipped with a C18 TSK-gel NPR (4.6 × 35 mm) column (Tosoh Bioscience, Montgomeryville, PA) while absorbance of the molecules eluting from the column was monitored between 220 and 400 nm with a UV-visible diode array detector (DAD, Model 335). The Star Chromatography workstation software (6.41 software, Agilent- Varian, Mississauga, Ont, Canada) controlled the respective outflow of 2 solvent pumps (model 210) drawing in eluent A (0.1% TFA in ACN) or B (0.1% TFA in 5% ACN: 95 % H2O) so that the mobile phase composition, initially set at 15% of eluent A, was ramped to reach a level of 65% at 6 minutes after injection. Software also controlled the auto sampler (Model 410) and a fraction collector (Model 701), displayed the absorbance at 220 nm, in addition to storing the three-dimensional data matrix from the DAD in the time, absorbance and wavelength domains. These files may be accessed with Polyview (Agilent-Varian version 6.41), a spectral processing application that allows to extract a spectrum from any point in time within a chromatogram, including at peaks, or to generate an absorbance chromatogram at any wavelength sampled by the DAD.
Three kinds of control runs were done regularly in order to detect problems related with the column, the extraction process or with the injection itself. Figure 1 shows that for each control, the absorbance at 220 nm increases slightly over time because of the continuous change in solvent composition during gradient elution. In blank runs (blue solid line), nothing is injected, but the column is exposed to a mobile phase identical to the one encountered during a run, enabling the detection of ghosting. This phenomenon is due to the subsequent elution of a protein previously bound to the column, thereby cross-contaminating successive chromatograms. Figure 1 also shows two other kinds of control: injection of a similar volume of initial mobile phase (green dot-dash) or injection of an extract from an unworn contact lens (pink dotted line). There were no peaks occurring in any of these control runs, except for the one associated with the dead time, which occurs at around 30 seconds. Extracts from worn contact lens and solutions containing known amounts of lysozyme or solutions of control and heated lysozyme were also injected in order to calibrate the intensity of the signal to the lysozyme level (see below), or the effect of thermal denaturation on peak parameters or elution profile.

Figure 1: Time-dependent absorbance (220 nm) of chromatographic control runs after real or simulated injections into the HPLC system. Controls show the observed chromatograms following the injection of: 1) initial mobile phase not exposed to a contact lens (green dot-dash) and 2) an extract from an unworn contact lens (pink dashed line). Blue solid line is the detector output during a control to detect ghosting (same gradient applied to the column, but without any injection).
Collection of tears and HPLC fractions and Western blots (trial run)
In trial runs, concentrated contact lens extracts were injected into the HPLC in order to test for the presence of lysozyme into the collected fractions. Fractions were collected between either 2.5 or 4 minutes (fraction CL1) or between 4 and 5.5 minutes (fraction CL2) of the chromatographic run. Collected fractions from 4 different contact lens extracts were pooled together, concentrated by centrifugation (1620 x g, 4°C, 135 minutes) through a centrifugal filter (Centriplus YM-10; Millipore), and assayed for protein concentration with bicinchoninic acid at 562 nm (microBCA, Pierce, Rockford, IL). The tears from 10 healthy volunteers were collected using 10 μl sterilized flamed disposable glass microcapillaries (Drummond, Broomall, PA). Tear samples were pooled together, frozen at -80°C until usage for Western blots or for separation of protein by size-exclusion (SE) chromatography on a TSKgel G3000SW 7.5 mm x 30 cm column (Tosoh Bioscience) [30].
Tears diluted to 1/8, concentrated fractions from tears or from contact lens extracts collected during SE- or RP-HPLC were separated by SDSPAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) on a 12% acrylamide separating gel with a Mini-Protean 3 cell (Bio-Rad, Hercules, CA). Proteins were transferred to nitrocellulose membrane overnight at 10 volts (4°C) on a Mini Trans-blot electrophoretic transfer cell under standard conditions. The membrane was then blocked with a solution containing 5% skim milk, exposed to an anti-human lysozyme (developed in sheep; Calbiochem-Millipore, Etobicoke, Ontario, Canada) at 1/100, and then to an anti-sheep IgG peroxidase. The membrane was revealed by chemiluminescence with ECL (Enhanced chemiluminescence reagents, GE Healthcare-Amersham, Mississauga, ON, Canada) on Kodak X-Omat films.
Calibration of lysozyme, analysis and statistics
Calibration experiments were repeated in order to ensure the obtention of a function that represented the series of experiments. The injection of external standards containing various amounts of lysozyme solubilised into extraction solution or initial mobile phase induced a chromatographic peak between 4.5 and 5.1 minutes. Data, stored in Excel spreadsheets, were transferred to SPSS (version 22.0.0.0) for statistical testing. First, standard least squares procedures were used in order to obtain distinct simple line equations for extraction solution (ES) for regular contact lens extracts or for concentrated extracts in the initial mobile phase (IMP) of peak area (PA) as a function of the mass of injected lysozyme (L). Figure 2 presents regression lines obtained at 220 nm depending on whether the solvent is the extraction solution (solid trace, PA=7.65 × 106* L+2.25 × 107, r2=0.98, p<0.000) or the initial mobile phase (dot-dash, PA=5.38 × 106* L + 7.01 × 106, r2=0.96, p<0.000) of the run.

Figure 2: Linear regressions between peak areas of lysozyme in chromatograms as a function of the injected mass. Lysozyme was dissolved into the initial mobile phase or into extraction solution. Both calibrations were obtained at 220 nm.
Differences between slopes and intercepts between these 2 line regressions were next tested for statistical signification using multiple linear regressions. This regression model includes the dichotomous dummy variable D coding for the treatment of contact lens extract either the regular extract in the extraction solution (in ES, D=0) or, the enriched extract into the initial mobile phase (in IMP, D=1). An interaction term between D and L is also included in this regression model [31]:
PA=α0+α1D+α2Lj+α3(DLj)+μj [32]
α0 and α2: intercept and slope for conditions of enriched extract into IMP, respectively;
α1 and α3: differential intercept and slope for conditions of regular extract into ES, respectively;
μj: error term.
The statistical significance of α1 and α3 implies that the intercept and slopes for ES and IMP are significantly different because both coefficients represent the differences between the 2 conditions in the intercept or in the slope [31]. When tested, the difference in the slopes was highly significant (p<0.000), but not the one between line intercepts (p=0.064). A peak-sensing routine in Polyview was applied to each chromatogram, and the amount of lysozyme was interpolated with the appropriate linear equation of Figure 2 once baseline absorbance had been corrected for changes in solvent composition during gradient elution runs.
Unmet assumptions about the distribution of data required for parametric tests required the use of non-parametric tests. The difference in median lysozyme levels between original and enriched samples was tested for statistical significance (p<0.05) with the Wilcoxon signedrank test for matched-pairs. The percent increase in detection (PID) of lysozyme in enriched samples over regular ones is obtained as follows:
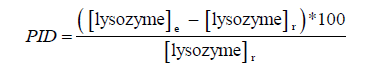
Where, (lysozyme)e is the average level of lysozyme measured in chromatograms obtained in enriched samples and (lysozyme)r is the average level of lysozyme measured similarly in samples with the regular extraction solution.
However, a correction is required when assessing how well both methods agree in the lysozyme content of the original extract. The quantity of lysozyme in the enriched extract was corrected for different injection volumes (10 vs. 50 μL) and concentration factors (8x vs. 1x) by multiplying the amount assayed in the concentrated aliquot by 0.625 (5/8).
We used Polyview to visualize absorbance spectra recorded with the DAD array. By observation of absorbance spectra at peak apex and at both ascending and descending inflection points, it is possible to assess the purity of an eluting component. An average purity parameter (PuP) was calculated for the apex of the peak and for the inflection points using Ai, the absorbance at wavelength λi:
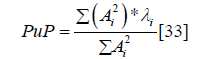
Correlation coefficients (similarity index) between these spectra were also calculated over the range of 220-400 nm. Stated otherwise, the similarity index is a measure of the sine of the angle between 2 spectra. Dissimilarity, the cosine of this angle, gives a better purity comparison when spectral differences are minimal. Spectral matching was done by comparing the normalized spectrum of the component eluting from lens extracts to the spectrum observed during calibration experiments with lysozyme (50 μM). Spectral searches were conducted across a 220-400 nm range by calculating a purity parameter for a suspected lysozyme peak and searching for a spectrum with a purity parameter that was ± 0.50 of the peak and with a coefficient of dissimilarity smaller than 0.03 [33,34].
Thermal denaturation experiments
A solution of lysozyme (0.2 mg/ml) was divided into 2 aliquots of equal volume. The first aliquot was heated for 2 hours at 100°C and the second, left untreated to serve as a control. In other similar, but separate experiments, an aliquot of a similar lysozyme solution was heated instead at 80°C. Both heated lysozyme solutions returned to room temperature before injection. Volumes of 10 μL of control and heated solutions of lysozyme were injected using a partial loopfill mode. In order to compare differences in peak areas between heated and control lysozyme, we tested whether or not the ratios of peak areas of heated lysozyme over areas of unheated lysozyme were different from 1 with the Wilcoxon signed-rank test for one sample. This test was selected because unmet assumptions precluded the use of parametric tests.
As shown by the Western blot of Figure 3, diluted (1/8) tear samples, pooled fractions of contact lens extracts collected between 4 and 5.5 minutes (CL2) in RP-HPLC and fraction of tears (F5) collected between 25 and 30 minutes during Size-Exclusion (SE)-HPLC reacted to lysozyme antibody. This retention time is expected in a SE-HPLC protocol for lysozyme to elute [30]. The antibody against lysozyme was very specific, as shown by a unique banding at the molecular weight expected for lysozyme. Lysozyme (~13 KD) is present in the fraction that elutes between 4 and 5.5 minutes (CL2), but not in fraction CL, eluting between 2.5 and 4 minutes (not shown).

Figure 3: Western blot confirmed that the same anti-lysozyme antibody reacted against a protein of about 10 kDa of MW (molecular weight) present in tears, in the (CL2) fraction from contact lens extracts eluting between 4 and 5.5 minutes and in the F5 fraction from tears eluting between 25-30 minutes on a size exclusion column.
Purity analysis and lysozyme levels in enriched and regular contact lens extracts
We proceeded next to compare the chromatograms obtained after injection of extracts from the same contact lenses but either left in the regular extraction solution or concentrated into a solvent identical to the mobile phase present at the time of injection into the column. Figure 4A presents 2 chromatograms obtained with extracts from CL-34, a methafilcon A-based contact lens. The pink dot-dash and the blue solid lines represent the changes in absorbance at 220 nm after the injection of 10 μl of the enriched extract and 50 μl of the regular extract, respectively. Lysozyme peaks occurred around 4.5-4.8 minutes. The percent coefficient of variation of the retention time averaged 0.6 ± 0.3 and 0.8 ± 1.2 % for those injections of the initial mobile phase and extraction solvent in Table 1. By observation of the Figure, the area under the peak is the largest upon injection of the enriched extract despite a volume of injection 5 times smaller than the one of the regular extract.
| aExtracts from the same contact lenses | ||||||
|---|---|---|---|---|---|---|
| Enriched Extracts in Initial Mobile Phase | Regular Extracts | |||||
| bExtract from contact lens number, Polymer (FDA group) | Days of wear | Lysozyme content (µg) | Lysozyme content (µg) | |||
| Measured | Corrected | Retention Time (RT) | Measured | RT (min) | ||
| CL-34, Methafilcon A (IV) | 15 | 44.7 ± 6.9 (5) | 27.9 | 4.6 ± 0.03 | 26.9 ± 21.8 (2) | 4.7 ± 0.1 |
| CL-35, Etafilcon A (IV) | 30 | 39.8 ± 3.2 (4) | 24.9 | 4.6 ± 0.01 | 26.8 ± 0.09 (3) | 4.7 ± 0.01 |
| CL-36, Alphafilcon A (II) | ? | 42.9 ± 4.3 (4) | 26.8 | 4.7 ± 0.04 | 21.5 ± 0.9 (3) | 4.7 ± 0.03 |
| CL-37 Alphafilcon A (II) | ? | 38.0 ± 6.1 (5) | 23.8 | 4.7 ± 0.03 | 19.3 ± 1.7 (2) | 4.7 ± 0.00 |
| CL-38 Balafilcon A (SHIII) | ? | 5.9 ± 2.3 (5) | 3.7 | 4.8 ± 0.04 | c0.9 (1) | 4.9 |
| Mean ± SEM | 34.2 ± 7.2 [39.8] | 21.4 | 4.7 | 19.1 ± 4.7 [21.5] | 4.7 | |
Table 1: Comparative lysozyme levels between aliquots of enriched and regular extracts from the same contact lenses.

Figure 4: Chromatogram of concentrated and regular extracts of lysozyme from the same contact lens and UV spectrum of lysozyme. A. Chromatogram obtained after injections of the following extracts from the same contact lens: 10 μl of the enriched extract dissolved into initial mobile phase (pink dot-dash), 50 μl of the regular extract sampled at 220 nm (blue solid trace). By inspection, the area under the peak of the enriched extract is larger than the one of the regular extract. Retention time (minutes) at peak apex is indicated for each chromatogram. B. UV Spectra insread of Absorbance spectrum at the peak of lysozyme obtained from the worn lens CL-36.
Table 1 presents the contact lens polymers from which lysozyme was extracted for the 5 ones with the highest lysozyme content. The wearing time, when available, the levels of lysozyme as well as the column retention time are also presented for extracts from the same contact lenses, but processed differently after extraction. Injections of 10 μl of enriched extracts led to a detection of a mean of 34.2 ± 7.2 μg (range: 5.9-44.7; median: 39.8 μg) of lysozyme whereas injections of 50 μl of the regular extract from the same contact lenses yielded an average of 19.1 ± 4.7 μg of lysozyme (range: 0.9-26.9; median: 21.5 μg). Differences between medians were significant (p=0.04) with the signed-rank test for matched pairs. In order to evaluate the agreement between estimates obtained with each protocol, the calculated amount of lysozyme corrected for the enrichment step (8 times more concentrated) and for the smaller volume injected (5 times less) is indicated in the table. As may be observed, there is not much difference between corrected levels of the modified protocol and those of the extraction solvent when lysozyme is abundant. However, when an extract contained less lysozyme, the peak surface area of the enriched extract is more likely to be detected compared to the regular extract from the same contact lens. This is the case of Cl-38, a silicone hydrogel contact lens. On 2 separate injections of the regular extraction solution from this lens (Table 1), lysozyme could be detected only in one chromatogram. In the second chromatogram, there was a small peak at 5.1 minutes, not identified as lysozyme by the peak sense function. This peak was also too small to be calibrated. Enrichment of the extraction solution from that lens led to positive detection of lysozyme due to sufficient amount of lysozyme that increased the signal and allowed its proper calibration.
On average, enrichment of the enriched extract led to a percent increase in detection of 79% compared to injection of regular extracts from the same contact lenses. As shown by Table 1, Methafilcon A and Etafilcon A contact lenses, CL-34 and 35, are among the 5 contact lenses with the highest lysozyme content per injection. These hydrogels are from group IV of the FDA classification: a highly hydrated polymer with ionic charges on their surfaces. Polymers from this group are known to bind much protein [7,8,11], and especially lysozyme [17,20,35,36], a positively charged protein in physiological conditions [6]. Alphafilcon A, a group II p-hema-based polymer, also contained much lysozyme [37]. However, the corrected lysozyme levels within the enriched extract are consistent with the measured levels of regular extracts from the same contact lenses.
Figure 4B uses the multi-wavelength information of the diode array detector in order to plot the absorbance spectrum observed at the apex of peak elution of lysozyme at 4.7 minutes for CL-36, an alphafilcon-A polymer. The profile of absorbance, maximal at 220 nm indicates that sampling at this wavelength is optimal. The absorbance spectrum observed at the peak of elution of a lysozyme standard (not shown) superimposed almost exactly over the one of CL-36. As shown by Table 2, the purity parameter (PUP) calculated for lysozyme peaks of chromatograms of contact lens extracts were very close to the one of the lysozyme standard (227.7). The library search feature of the software identified the lysozyme standard to match the peak eluting in the contact lens extract, suggesting once again that lysozyme is the eluting species. The table also presents the similarity and dissimilarity coefficients between the spectra across the peak and the purity parameter for the same extracts analyzed in Table 1. The purity parameter across a peak did not change. This indicates consistency of its spectral characteristics across the peak and suggests that the peak is essentially free from contaminants [29,34].
| aExtracts from the same contact lenses | ||||||
|---|---|---|---|---|---|---|
| Enriched Extracts in Initial Mobile Phase | Regular Extracts | |||||
| Lysozyme standard | Similarity | Dissimilarity | PuP (nm) | Similarity | Dissimilarity | PUP (nm) |
| Lysozyme (2.5 µg) | 0.999 ± 0.0001 (8) | 0.006 ± 0.005 | 227.77 ± 0.09 | 0.999 ± 0.000 (4) | 0.011 ± 0.001 | 227.72 ± 0.04 |
| eExtracts from CL No, Polymer | ||||||
| CL-34, Methafilcon A | 0.999 ± 0.000 (5) | 0.010 ± 0.006 | 228.01 ± 0.18 | 0.999 ± 0.000 (2) | 0.018 ± 0.012 | 227.97 ± 0.33 |
| CL-35, Etafilcon A | 0.999 ± 0.000 (4) | 0.010 ± 0.005 | 227.97 ± 0.11 | 0.999 ± 0.000 (3) | 0.010 ± 0.002 | 228.05 ± 0.12 |
| CL-36, Alphafilcon A | 0.999 ± 0.000 (4) | 0.012 ± 0.001 | 228.02 ± 0.04 | 0.999 ± 0.000 (3) | 0.010 ± 0.003 | 227.99 ± 0.19 |
| CL-37, Alphafilcon A | 0.999 ± 0.000 (5) | 0.011 ± 0.003 | 228.13 ± 0.09 | 0.999 ± 0.000 (2) | 0.010 ± 0.002 | 227.89 ± 0.05 |
| CL-38, Balafilcon A | 0.999 ± 0.000 (5) | 0.005 ± 0.003 | 227.64 ± 0.07 | 0.999 (1) | 0.009 | 227.69 |
| Mean ± SEM | 0.999 ± 0.000 | 0.009 ± 0.001 | 227.96 ± 0.082 | 0.999 ± 0.000 | 0.011 ± 0.002 | 227.92 ± 0.063 |
Table 2: Spectral matching, purity analysis of contact lens extracts.
The effect of heating lysozyme on peak area and elution profile
As shown in Figure 5A, heating lysozyme at 100°C changed much the shape of the eluting peak, which appeared much broader but less high than its control. Presumably, this indicates changes to the protein. At 80°C, changes were more subtle as shown by the appearance of control (dotted trace, left) and heated lysozyme (dashed trace, right) in Figure 5B. The chromatogram representing heated lysozyme appeared less symmetrical compared to control but presented, as for lysozyme heated at 100°C, a front shoulder (blue empty arrow) and an additional inflection point (purple arrow), both of which are located on the uprise before the peak (dashed arrow). At the higher temperature, this additional inflection point is moved further away from the apex (Tables 3 and 4: 0.34 min before the apex at 100°C and 0.14 min before the apex at 80°C). The changes observed in these figures are typical and were consistently revealed by the peak sensing function of Polyview in chromatograms of heated lysozyme, but never appeared in chromatograms of control unheated lysozyme solutions or in contact lens extracts. Figure 5C presents the absorbance spectrum at the apex of lysozyme control (lower blue trace) and in different positions across the peak of the lysozyme solution heated at 100°C shown in Figure 5A. The 3 traces of heated lysozyme indicate a shift in the absorbance spectrum at the peak apex (red trace) and across, especially visible around 250 nm. The spectra near the front shoulder (green trace) and the downslope inflection points are even more modified.
| Heated lysozyme (100°C) | Control | ||||||
|---|---|---|---|---|---|---|---|
| No | tR (min) | Spectrum | PuP (230 ->300 nm) | No | tR (min) | Spectrum | PuP (230->300 nm) |
| 1 | 4.658 | PeakApex | 248.980 | 1 | 4.697 | PeakApex | 249.090 |
| 2 | 4.603 | aUpslope | 248.776 | 2 | 4.643 | Upslope | 248.666 |
| 3 | 4.702 | DownSlope | 249.012 | 3 | 4.742 | DownSlope | 248.761 |
| 4 | 4.492 | FShoulder | 248.627 | ||||
| 4.317 | 2nd inflection pt | 248.390 | |||||
| Statistics | Statistics | ||||||
| Best Correlation: 1 and 3; bSim: 0.999998; Dissim: 0.001983 | Best Correlation: 2 and 3; Sim: 0.999992; Dissim: 0.003937 | ||||||
| Worst Correlation: 1 and 4; Sim: 0.999170; Dissim: 0.040739 | Worst Correlation: 1 and 2; Sim: 0.999946; Dissim: 0.010346 | ||||||
| Average PuP=248.849 nm; Standard Deviation=0.157 nm | Average PuP = 248.839 nm; Standard Deviation = 0.182 nm | ||||||
Table 3: Purity parameter (PuP) in UV Spectra of control and experimental (heated at 100°C) lysozyme solutions.
| Heated lysozyme (80°C) | Control | |||||||
|---|---|---|---|---|---|---|---|---|
| No | tR (min) | Spectrum | PuP (230 ->300 nm) | No | tR (min) | Spectrum | PuP (230->300 nm) | |
| 1 | 4.750 | PeakApex | 248.875 | 1 | 4.752 | PeakApex | 248.726 | |
| 2 | 4.705 | aUpslope | 248.521 | 2 | 4.703 | Upslope | 248.260 | |
| 3 | 4.797 | DownSlope | 248.683 | 3 | 4.797 | DownSlope | 248.387 | |
| 4 | 4.633 | FShoulder | 248.734 | |||||
| 4.612 | 2nd inflection pt | 248.681 | ||||||
| Statistics | Statistics | |||||||
| Best Correlation: 2 and 3; bSim: 0.999989; Dissim: 0.004721 | Best Correlation: 2 and 3; Sim: 0.999992; Dissim: 0.004012 | |||||||
| Worst Correlation: 1 and 4; Sim: 0.999887; Dissim: 0.015006 | Worst Correlation: 1 and 2; Sim: 0.999936; Dissim: 0.011310 | |||||||
| Average PuP = 248.703 nm; Standard Deviation = 0.127 nm | Average PuP = 248.458 nm; Standard Deviation = 0.197 nm | |||||||
Table 4: Purity parameter in UV Spectra of control and experimental (heated at 80°C) lysozyme solutions.~

Figure 5: Effect of heating on the elution profile and action spectrum of lysozyme. A. Chromatograms of absorbance at 220 nm obtained upon the injection of a solution of 0.2 mg/ml lysozyme either left at room temperature (red trace) or previously heated at 100°C for 2 hours (blue trace). The times at which the absorbance spectrum is viewed in C is shown by short dotted lines crossing the chromatogram of heated lysozyme: green, 4.405 min; red, peak apex at 4.658 min; orange, 4.847 min. Blue dotted line indicates the apex of the peak of control lysozyme at 4.697 minutes. B. Magnified chromatograms in the area of the peak of a control solution of lysozyme (dotted trace, left) and one previously heated at 80°C (dashed trace, right). As indicated by the peak sensing routine, the chromatogram of heated lysozyme shows an additional inflection point (purple arrow) and a shoulder (blue empty arrow) before the peak (dashed arrow) not observed in the control chromatogram. C. Normalized absorbance spectra of lysozyme heated at 100°C, obtained at the times indicated in A for the green, red and orange traces present differences with the one of control lysozyme (blue trace) especially in the 240-260 nm range.
Tables 3 and 4 present the coefficient of purity across the peak for the chromatograms shown in Figure 5 for temperatures of 100 and 80°C. At both temperatures, the correlation of absorbance spectra was the worst between the apex and the front shoulder, representing dissimilarity coefficients of 4.1 and 1.5% at 100 and 80°C, respectively. These coefficients are larger than those found for contact lens extracts reported in table 2 (≈1%). Finally, median ratios of heated lysozyme peak area over control peak area were respectively 0.908 (n=22) and 0.947 (n=28) for heating temperatures of 100 and 80°C. This ratio was significantly different from 1 (p=0.022) at the temperature of 100°C, but not at 80°C, as indicated by separate Wilcoxon signed-rank tests. Therefore, heating lysozyme to 100°C reduces peak size. Changes induced by heating lysozyme at 80°C or higher are detectable by modification of the absorbance spectrum, and by an alteration of the profile of the peak.
We amplified the HPLC signal of lysozyme extracted from ex vivo contact lenses [5]. The analysis of the fractions collected in trial chromatographic runs revealed that lysozyme eluted between 4 and 5.5 minutes, as indicated by their positive reaction with an antibody against lysozyme. The post-processing capabilities of the DAD array allowed including a range of wavelength in the analysis of experimental chromatograms in order to obtain relative absorbance spectra. Such analyses allowed to 1- compare peak absorbance spectra of contact lens extracts to lysozyme standards, and, 2- observe absorbance spectra across peak area to determine peak purity. All of these procedures pointed to lysozyme as the species eluting after its extraction from worn contact lenses of different brands.
Rather than monitoring the deposits formed on one or two brands of contact lenses, either exposed to an in vitro solution of artificial tears or fitted to a clinical sample of patients, we tried to take advantage of our university clinic by requesting volunteer student clinicians to anonymously collect lenses of any brand left by their consenting patients disposing of their used lenses. Because of the anonymity, neither the type of lens nor the extent of wear could be controlled or verified. Under these conditions, there could be no control over material type, duration of wear, care system or handling by patients. Therefore, the data presented herein should not be used to compare contact lens materials as to how they attract lysozyme. It is rather useful to compare the measured and corrected levels of lysozyme in the regular and enriched extracts, in order to assess the merits of concentrating the extracts to amplify the absorbance signal.
In a first part, we compared the estimates of lysozyme content from the same contact lens but either left in the original extraction solution or concentrated into the initial mobile phase. When corrected for the different concentration factor and volume of injection, the estimated amount of lysozyme in concentrated extracts was in agreement with the one of the regular extracts obtained from the same lenses. Concentrating the contact lens extract results in a 79% increase in detection level compared to the original extract. Such an increase represents a marked improvement, which could mean difference between peaks that are well separated and others that are not. At a low concentration of lysozyme, it is more likely that random error will be responsible for the nondetection of a peak or insufficient peak size to allow calibration. This occurred in the injection of a regular extract from a silicone hydrogel contact lens: in one injection, lysozyme could be detected whereas; it could not be detected in another injection of the same extract. Upon injection of the enriched extract from the same contact lens, lysozyme was unambiguously identified at all times.
Increased amplification of the signal is also visible in Figure 4A by comparing the larger peak areas of enriched contact lens extracts compared to regular ones. This increased sensitivity is certainly related to increased chemical activity (concentration) of lysozyme in the enriched extract, and, for specimens of low concentration, with the obtention of a critical mass detectable by HPLC. It is also possible that solubilisation of the enriched extract in a solvent similar in composition to the mobile phase initially pumped into the system contributes to this increased sensitivity. Less unpredictable chemical interactions should occur when the solvent composition of the injected extract and the mobile phase are identical.
The herein proposed method of lysozyme quantitation requires a few extra steps to the original method. However, these short steps increase its sensitivity. The use of a diode array detector added extra support to the elution of lysozyme at about 5 minutes and allowed to match its absorbance spectrum with the one of an external lysozyme standard. Observation of spectral absorbance, coefficient of similarity and purity parameter across the eluting peak suggested that the peak was pure. Whereas concentrating the extract makes it easier to detect peaks, it is done at the expense of a reduced volume of extract, and therefore, reduced possible number of repeated chromatograms from the same extract.
It is likely that lysozyme does not maintain its native conformation during its elution in this protocol, as occur usually in separations by RP-HPLC. However, proteins smaller than 20 kDa can often be renatured after separation [38]. An indication from the literature suggests that denatured lysozyme could be related to lens discomfort [19]. Heating lysozyme has been used to study its denaturation in various biochemical assays [39] or on contact lenses [15]. Despite that denaturation of lysozyme encountered in situ during contact lens wear is probably different from the one encountered by a thermal treatment, we heated lysozyme in order to determine whether or not this protocol of RP-HPLC could detect lysozyme denatured by a thermal treatment. We show that heating lysozyme to 80°C is sufficient to affect its elution profile on this chromatographic column. Hall et al. have shown the absence of any active lysozyme on contact lenses heated to 80°C during 2 hours [15]. Heating lysozyme to 100°C for 2 hours, known to irreversibly affect lysozyme function [39], influences peak size. Heating lysozyme dramatically affects the shape of the peak of the eluting protein and its absorbance spectrum. The changes encountered when lysozyme was heated were never observed on chromatograms after the injection of contact lens extracts.
In summary, the identity of lysozyme has been confirmed by a typical retention time during chromatography, by binding of the eluting protein and of tear samples to the lysozyme antibody in the Western blot, and by a similar absorbance spectrum of the eluting compound compared to a lysozyme standard. A step of enrichment of the protein extract coupled with solubilization into the initial mobile phase can be useful to improve sensitivity of this HPLC method in order to detect a level of lysozyme otherwise below the sensitivity of the measurement. Further increases in the enrichment ratio could be useful to decrease the limit of detection of this method. This protocol is able to detect denaturation of lysozyme induced by heating at 80°C or higher.
The HPLC used in these experiments was obtained by funding provided by the Canada Foundation for the Innovation. No other financial support was obtained for the conduct of the research or preparation of the article. Langis Michaud has received lectureship honoraria from Bausch & Lomb, Cooper Vision, and research funds from Johnson & Johnson Vision Care. The authors do not have any financial interest or conflict of interest in any of the products or processes mentioned in this manuscript. The authors acknowledge expert technical assistance of Ms Elodie Gorju, Assia Abouchita and Radostina Ratchkova. Thanks to Ms Micheline Gloin for the artwork.