Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2014) Volume 7, Issue 8
A genomic island (GI) is a genomic segment in a host genome, and it was transferred from donor genomes. Since genomic islands (GIs) usually contain important genes such as pathogenicity genes, the detection of GIs becomes extremely critical to medical research and industrial applications. Previous computational GI detection tools used one or a few GI-associate features, and thus they suffered the problem of low prediction accuracy. A systematic approach that uses multiple sources to improve GI prediction accuracy, therefore, is in great demand. In this paper, we report the development of Genomic Island Hunter (GIHunter), an accurate software tool for GI detection. GIHunter is a decision tree based bagging model that uses eight GI-associated features such as sequence composition, mobile gene information, and integrase. The performance metric comparison between our approach and other current existing GI prediction methods has shown that our approach is more accurate than other approaches. We have used GIHunter to predict GIs in more than 2000 prokaryotic genomes. We have also visualized our predicted GIs so that our predicted results become useful and meaningful for biomedical studies. Our GI program GIHunter can be obtained at: http://www.esu.edu/cpsc/che_lab/software/GIHunter. Our GI prediction results are available on our genomic islands database, which is: http://www5.esu.edu/cpsc/bioinfo/dgi.
Keywords: Genomic islands, Machine learning, Genomes, Bacteria, Archaea, Computational methods
Genomic Islands (GIs) are segments of genomic sequences that were horizontally transferred from other bacterial species or viruses [1]. The driving forces of the forming of GIs in such genomes could be attributed to the adaptability to their ever changing surroundings, and thus the contents of GIs in the host genomes could be versatile [2]. For example, some GIs are pathogenic-related, while some others may confer drug-resistance to bacterium. Some GIs contain genes which could produce secondary metabolites, while some others degrade recalcitrant chemical products.
It is extremely significant to identify the locations and contents of GIs because such findings will be a greatly benefit to biomedical research. For instance, we can design and produce corresponding antibiotics based on the identification of pathogenic GIs [3]. On the other hand, for those GIs containing beneficial secondary metabolites, we can produce them on a large scale [4]. Due to the explosive growth of genomic sequence data, huge amounts of genomic structure information need to be elucidated. Unfortunately, biological experiments may only contribute us a small fraction of information for GIs in all sequenced genomes. Therefore, it is urgent to develop computational tools to identify all GIs.
Currently, there are mainly two kinds of GIs detection approaches: comparative genomic-based approach, and sequence compositionbased approach [5]. Comparative genomic-based approach compares the query gene sequence with its closely related species genomes by aligning them together and considering the genome segments that are only present in the query genomic sequence to be GIs [6]. Some examples of tools based on this approach include IslandPick [6] and MobilomeFinder [7]. While the detection of GIs using this kind of approach is relatively reliable, it is limited to a small group of genomes. The reason is that not all genomes have an enough number of closely related genomes for references, and thus they cannot be analyzed by using this approach. Furthermore, such tools also need manual adjustment and selection, which is hard to perform and control because it may lead to inconsistent selection criteria due to the unfamiliarity of different genome structures [8].
Sequence composition-based approach is based on the general concept that different species have different genomic sequence signatures. Thus, if there is a genomic region that has special signature different from that of the majority regions of the genome, this region is considered to be a GI. Accordingly, this kind of approach enables us to detect GIs within a genomic sequence without relying on reference genomes or manual selections, and thus it can be applied to any sequenced genome.
The key of this kind of approach is to detect abnormal sequence compositions or signatures in the genome. Such signatures include codon usage [9], and G+C content [10]. Some GIs are also flanked by tRNA genes [11]. Furthermore, some others GIs could contain mobile genes such as integrase and transposes [12]. Computational tools using this kind of approach include AlienHunter [13], Centroid [14], IslandPath [15], PAI-IDA [16] and SIGI-HMM [17]. Recently, some integrative approaches uses the prediction results of individual programs to obtain census GIs. Available programs and tools include EGID [18], GIST [19] and IsalndViewer [20].
Despite of the availability of existing GI prediction tools, the prediction accuracy is not as good as desirable. The causes of the low prediction accuracy are due to the amelioration of genomes, which led to false negatives [9], or the existence of abnormal sequence composition such as ribosome regions in the host genome, which leads to false positives. Therefore, it may not be sufficient to use only one or two features to determine a sequence is a GIs or not. Another problem for GIs detection is the boundary of GIs. Some GIs only cover several kilo bases (kbs) while others can cover hundreds of kbs, which makes it difficult to determine the size of GIs.
In this paper, we propose an accurate GI prediction approach that incorporates multiple features, including: 1) sequence composition based feature, Interpolated Variable Order Motifs (IVOM); 2) mobile gene information, Integrase, and transposases; 3) tRNA gene; 4) phage information, and 5) two new features we found recently, intergenic distance and highly expressed genes (HEGs). We use decision-tree based ensemble learning to build the GI model based on these features. Additionally, in order to resolve the GI boundary issue, we treat each gene in GIs individually, and predict GI gene or non-GI gene by using feature values associated with its neighboring region, and then piece contiguous GI genes together using our GI gene merging process to predict GIs. The experimental results have shown that our approach has outperformed other GI tools in terms of prediction accuracy, suggesting the power of our approach, and its potential use in the future GI prediction.
Computational framework
Our computational framework for genomic island prediction was divided into two main stages: (1) model construction for GI/non-GI gene classification, and (2) genome scale genomic island prediction.
The stage of building GI/non-GI gene classifier model consisted of the following steps:
1. Collecting training data and genomic sequence data. The training dataset of known genomic islands and non-genomic islands were collected for model construction. The genomic sequence was collected for feature value extraction.
2. Extracting genomic island-related features. Feature values for all GI and non-GI genes from the training data were computed.
3. Building a GI/non-GI gene classifier model. The feature values obtained in Step 2 were used to build a model for classifying any gene to be either GI or non-GI.
The stage of genome scale genomic island prediction consisted of the following steps:
1. Collecting genomic sequence. The genomic sequence to be analyzed was collected, and was used for feature value extraction.
2. Extracting genomic island-related features. Feature values for all the genes in the genome were computed.
3. Classifying GI/non-GI genes using the model built from Stage 1. Based on the GI/non-GI gene classifier model, all genes in the genome were classified to be a GI gene or non-GI gene.
4. Predicting genomic islands based on classified genes. Our program merged the contiguous GI genes and predict them a genomic island.
The computational framework flowchart for genome scale genomic islands is summarized in Figure 1. The detailed procedures are presented in the following subsections.

Figure 1: Computational framework for the genome scale genomic island prediction.
Dataset collection
The positive genomic islands (GIs) and negative genomic islands (non-GIs) datasets used in this study were obtained from [6]. The dataset contains 771 GIs and 3770 non-GIs. They covered 118 genomes in total, and they were used for building decision-tree based GI prediction model.
The sequence lengths of original GIs and non-GIs varied from 8 kb to 31 kb, making it less meaningful using GI-associated feature values with different GI or non-GI sizes to build our prediction model. In order to resolve this problem, we extracted all genes within the training GI and non-GI datasets. From this training dataset, we obtained 10,459 genes from GIs, and 44,909 genes from non-GIs, with the total of 55,368 genes. Since these genes were from 118 genomes, representative specie genomes from the domains of Bacteria and Archaea. Thus, we believe that the selected genes in the training set should not be overly biased, and will not affect our model overall.
To obtain the feature values for GIs / non-GIs genes in the training dataset (or all genes in a genome used for genomic island prediction), we downloaded the corresponding complete genome sequences and the annotations from the National Center for Biotechnology Information (NCBI) FTP server (ftp://ftp.ncbi.nih.gov/genomes/Bacteria).
Feature value extraction
For each of 55,319 genes from GI and non-GI datasets, we calculated the GI-associated feature values. The calculation of feature values for any gene was not based on the gene itself. Instead, it relied on the gene and its neighboring region. In this study, we selected the region length of 8 kb because the feature information of a-8kb-long region can roughly reflect the existence of a genomic island or not. Figure 2 illustrates a selected gene in a genomic island, and its’ corresponding region (8 kb) for calculating feature values.

Figure 2:An illustration of extracting feature values for a gene within a GI. The box denotes a genomic island, where circles represent genes, and the black circle is the selected GI gene for feature value calculation. The feature value calculation is based on the dashed box, which is chosen by starting from the center of the selected gene, extending to left by 4 kb, and then extending to right by 4 kb.
For any gene, we calculated eight features, including IVOM, Density, Integrase, Phage, tRNA, Highly Expressed Genes (HEGs), Average intergenic gene distance (AvgID), and Transposase, based on its surrounding regions (8 kb). The reason of choosing these eight features was due to their contributions to genomic island prediction in previous studies [8,21,22].
We calculated each of eight feature values using the methods summarized in Table 1. Specifically, we used Alien Hunter [13] to obtain IVOM score for the selected gene within the genomic segment information of 8 kb (Figure 2). For the features of tRNA, Phage, Integrase, and Transposase, we used regular expression based search on annotated genome files information to count how many genes within the 8kb region that have these features. For the feature of HEGs, we considered ribosomal protein (RP) genes, translation and transcriptional processing factor (TF) genes (e.g., RNA helicase, RNA polymerase, tRNA synthetase), chaperone degradation (CH) genes, and genes involved in energy metabolism (e.g., NADH) to be highly expressed genes [23], and counted the number of HEGs within the 8kb region.
| Feature | Relation with genomic islands (GIs) | Feature value obtained |
|---|---|---|
| IVOM | GIs have higher IVOM scores (i.e., sequence composition bias) | AlienHunter |
| tRNA | GIs are flanked by RNA genes | Functional annotation |
| Phage | GIs may contain phage-related genes | Functional annotation |
| Integrase | GIs may contain integrase genes | Functional annotation |
| Transposase | GIs may contain transposase genes | Functional annotation |
| Highly expressed gene | GIs may not contain highly expressed genes | Functional annotation |
| Gene density | GIs have lower gene densities | Genome sequence |
| Average intergenic distance | GIs have higher inter-genic gene distance | Genome sequence |
Table 1: Summary of genomic island associated features.
For the feature of gene density, we counted the number of genes within the 8kb region, and thus gene density was defined as the number of genes per kb. For the feature of average inter- genic distance, we first measured inter-genic distances for each adjacent gene pair (gi, gi+1) within the 8 kb region, and then averaged them to obtain AvgID. The features of density and AvgID may not be as effective as the features of IVOM or mobility genes. Nevertheless, they have some differentiation power as shown in Supplementary Figure 1, and thus we believe they could improve differentiating non-GIs and GIs when adding these features. The feature value difference on non-GIs and GIs in the training dataset is shown in Supplementary Figure 1, and they have been summarized in Table 1.
Model for classifying GI/non-GI genes
Our genomic island gene model is a bootstrap aggregating (also known as bagging) of base classifiers, in which each base classifier is a decision trees in this study. Bagging [24] is a method whose classification takes the majority votes of multiple classifiers, with the same weight for each of base classifiers (Figure 3A). The Bagging model used in this study consists of multiple decision trees as base classifiers in the “committee”. In this study, we used WEKA [25] to build our decision tree based bagging model for GI gene prediction.

Figure 3:A decision-tree based bagging model for GI/non-GI gene classification. (A) A decision-tree based bagging classifier; (B) An example of decision tree based classifier, which is used in bagging in (A). The pie chart in the bottom shows the probabilities of being GI gene, colored in “Blue” or non-GI gene, colored in “Red”.
The training set used for constructing each decision tree was sampled by bootstrap sampling of 55,319 examples. The training examples are a set of tuples
The construction of a decision tree model was based on the ID3 algorithm [26], which was a top-down greedy search algorithm. It first started with the whole training set, and then chose the best feature as the root node based on the information gain values. It then split the set based on the possible values of the selected best feature. The algorithm assigned the node to a leaf node when all instances in a subset had the same classification, and labelled the same classification as the instances. For the mixed label classification, the algorithm continued to choose the next best feature based on the subset of the training examples. This whole process was repeated until no further features were available.
In this study, we used J48 in WEKA, which is the Java implementation of C4.8 algorithm [25]. C4.8 is the latest research version for the C4.5 algorithm, which is one of the best-known and most widely-used decision tree algorithms. C4.5 extends the ID3 algorithm by addressing several important issues. The C4.5 algorithm handles numerical attributes and missing values, it also incorporates post-pruning process to handle the over-fitting problem.
Genome scale genomic island prediction
The trained model built using J48 decision tree based ensemble algorithms were used to classify all genes (i.e., GI gene or non-GI gene) in any query genome. The inputs for GI prediction were: (a) the whole genome sequence; (b) the gene annotation of the genome. The procedure of calculating feature values for all genes in the genome is as same as that of described in the previous Section, i.e., 1) Finding the neighboring region corresponding to the selected gene; and 2) Calculating each of eight feature values (IVOM, Density, Integrase, Phage, tRNA, HEG, AvgID, and Transposase) for each selected region.
Based on the feature values for each gene in the genome, we classified these genes into either GI genes (or non-GI genes) using our decision-tree based bagging model built from the training set. Such GI gene (or non-GI gene) unit information was further processed, and we obtained genomic islands through the following steps:
1. Finding all GI segments based on the model. From the decisiontree based bagging model, each gene for the whole genome was classified into GI-genes or non-GI genes. We considered those contiguous GI genes in the genome to be a GI segment, and we found all GI segments.
2. Picking two neighboring GI segments, Gi and Gi+1, calculating the average probability value for the region spanning from the first gene of Gi to the last gene of Gi+1, including those non- GI genes between Gi and Gi+1. We merged smaller adjacent GI segments into a new GI segment when the average value was greater than a threshold value T. A larger GI segment of G2 in Figure 4 is the product of two neighboring GI segments merged. The optimal threshold value T was determined by selecting a set of values between 0 and 1, running on the benchmark sets, and comparing performance metrics of predicted GIs under different threshold settings. In this study, we found that T=0.6 was the optimal threshold, and chose it as the cut-off value.

Figure 4:The merging process of combining GI genes into GIs. Each bar represents a gene in the genome, with the bar height representing the probability of being a GI-gene, calculated from our bagging model. The GIs are basically the genomic regions with a cluster of adjacent GI genes as shown in G1, G3 and G4. Some adjacent GI gene clusters can be extended to become a single GI, as shown in G2.
3. Repeating the process of Step 2 until non neighboring GI segments could be merged;
4. Filtering out short GI segments. In this study, we removed those GI segments containing less than six genes. Figure 4 shows an example of a short GI segment between G1 and G2 removed. The remaining GI segments were considered to be final predicted genomic islands for the whole genome.
We have developed our program, Genomic Island Hunter (GIHunter), for predicting genomic islands in the whole genome, as described above.
Performance evaluation
We looked into the performance of our computational framework from two aspects: a) The accuracy of the decision-tree bagging model for GI/non-GI gene classification, and b) The prediction accuracy of predicted genomic islands.
Classification accuracy on GI/non-GI genes
We used a ten-fold cross-validation scheme to evaluate the GI/ non-GI gene classification accuracy on our ensemble learning model. In particular, the known GI/non-GI dataset was evenly separated into ten parts, and the first part was evaluated based on the model trained from the remaining nine parts. This process continued until all ten parts were evaluated. The overall classification accuracy was the average of all ten separate evaluations. True positives (TP) were the number of GI genes predicted to be GI genes. False negatives (FN) were the number of GI genes predicted to be non-GI genes. True Negatives (TN) were the number of non-GI genes predicted to be non-GI genes. False positives (FP) were the number of non-GI genes predicted to be GI genes. The recall, precision, and classification accuracy were defined as follows:
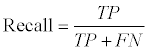 (1)
(1)
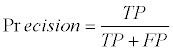 (2)
(2)
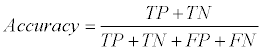 (3)
(3)
Genomic island prediction accuracy
We also compared our predicted GIs with the benchmark dataset. We evaluated our predicted GIs at the nucleotide level. In particularly, true positives (TP) were the number of nucleotides both in benchmark GI dataset and in predicted GIs. True negatives (TN) were the number of nucleotides both in benchmark non-GI dataset and predicted non- GIs. False positives (FP) were the number of nucleotides in predicted GIs but not in benchmark GI dataset. False negatives (FN) were the number of nucleotides in benchmark GI dataset but not in predicted GIs. We focused on four performance measures, recall (See equation 1), precision (See equation 2), Performance Coefficient (PC) (See equation 4), and F-Measure (See equation 5).
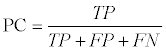 (4)
(4)
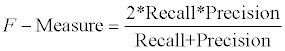 (5)
(5)
Classification accuracy on GI/non-GI genes
To evaluate the classification power of GI/non-GI genes, we applied the decision tree J48 model on training datasets. We used default parameter settings, with the confidence factor of 0.25 and minimum number of instance per leaf of 2. We have achieved the recall of 0.682, precision of 0.879, and the overall accuracy of 0.922. The classification results indicate that this decision tree model with eight features is quite specific (0.879), although it is not quite sensitive (0.682).
As a comparison, we also applied four other classifiers, including Naïve Bayes, logistic classification and regression trees (CART), and Support Vector Machines (SVMs), to the same datasets. These algorithms with default parameter settings were run through WEKA. As shown in Figure 5, the classification powers of the other four algorithms were slightly lower than that of the decision tree when such three performance measures (recall, precision and accuracy) were used for comparison. We also tested the J48 decision tree based bagging model in order to improve classification accuracy. Our classification results showed that this bagging model performed better than a single decision tree model, with the increase of 2.7% for recall, 0.6% for precision, and 0.6% for overall accuracy (Figure 5). Figure 6 shows ROC curves for decision tree model, and a decision tree that was built by J48 algorithm on the training set is shown in Supplementary Figure 2.

Figure 5: GI/non-GI gene classification accuracy with different machine learning algorithms.

Figure 6: The ROC curve for decision tree model in GIHunter.
Prediction accuracy on genomic islands
The measure of classification accuracy on GI/non-GI genes is an indirect way to evaluate the performance of our decision-tree based bagging model. To truly measure the prediction power of our approach for predicting genomic islands, we directly compared our predicted genomic islands with the benchmark dataset. To do so, we collected 118 prokaryotic genomes from the National Center for Biotechnology Information (NCBI) FTP server, ran our GIHunter (http://www5.esu.edu/cpsc/bioinfo/software/GIHunter) program, and generated GI locations for each genome. We used genomic islands obtained by IslandPick [27] as benchmark to evaluate the predicted GIs by GIHunter. We also collected predicted GI results of five component programs including AlienHunter, IslandPath, SIGI-HMM, INDeGeNIUS, and PAI-IDA.
As we can see in Figure 7, our program GIHunter reaches the highest recall (0.69), highest precision rate (0.91), highest PC (0.65), and highest F-measure (0.78), indicating the improvement of prediction accuracy of our method over previous approaches. The improvement of prediction accuracy is due to the integration of multiple sources (i.e., multiple features). Our program combines the IVOM score, generated by AlienHunter, with other GI-related information such as phage, integrase, transposase, tRNA, which were used in the programs of IslandPath and PAI-IDA. In addition, we also include new information such as HEGs, and inter-genic gene distance.

Figure 7:Performance comparison with different GI prediction tools.
The performance measures include recall, precision, nPc and F-measure. GI prediction tools include Alien hunter, IslandPath, SIGI-HMM, INDeGeNIUS, PAI-IDA and our program, GIHunter.
Case Study: predicted genomic islands in Corynebacterium diphtheria NCTC13129
C. diphtheria NCTC13129 is a microbe that produces diphtheria toxin (DT), which causes the symptoms of diphtheria [28]. It was first isolated from the Pharyngeal membrane of a 72-year-old female with clinical diphtheria [29]. The genome has been fully sequenced, with the size of 2,488,635bp. In this genome, we predicted fourteen genomic islands in total. The visualization view of fourteen genomic islands as well as eight feature values is shown in Figure 8. More detailed information about fourteen genomic islands is described in Table 2.
| Label | Start / Stop | Genes covered (total number of genes) | Supporting information |
|---|---|---|---|
| GI_1 | 21,381-390,836 bp | DIP0021 -> DIP0425 (404) |
This region contains two integrase genes annotated as phage integrase, five phage genes annotated as phage prohead protease, phage capsid protein, and phage tail fiber protein, ten tRNA genes and five transposon related genes encoding for IS element transposase. The overall IVOM score in this region is 18.19, significantly higher than that of in non-GI region (usually < 10). |
| GI_2 | 403,882 -421,292 bp | DIP0441 -> DIP0452 (11) | The IVOM score is 24.18, much higher than that of the rest of genomic region. This region contains one transposon related gene. This genomic island was supported by the genome analysis in [17], where researchers considered the gene region from DIP0438 to DIP0445 to be a pathogenicity island. |
| GI_3 | 460,848 -490,567 bp | DIP0495 -> DIP0521 (26) | The high IVOM score (17.98) in this region indicates sequence composition is different from that of its neighbor genomic regions. |
| GI_4 | 726,536 -799,536 bp | DIP0750 -> DIP0826 (76) | This predicted GI region contains one phage integrase gene, one tRNA gene, and six transposon related genes including transposase A and IS element transposase. IslandViewer also has one corresponding genomic island from DIP0806->DIP0822. Other research results also discovered two genomic islands, including DIP0752 -> DIP0766 and DIP0795 -> DIP0820, in this region |
| GI_5 | 929,413 -953,582 bp | DIP0957 -> DIP0987 (30) | The overall IVOM score in this region is 18.64. IslandViewer also predicted the genomic region from DIP0960 to DIP0966 to be a genomic island. |
| GI_6 | 1,136,731 -1,139,080 bp | DIP01150 -> DIP1158 (9) | The main evidence of predicted GI is due to its very high IVOM score (24.81) |
| GI_7 | 1,160,398 -1,699,168 bp | DIP1168 -> DIP1662 (494) | This region contains nine tRNA genes, and four putative transposase genes. IslandViewer predicted three GIs in this region, i.e., DIP1184 -> DIP1188, DIP1444 -> DIP1453, and DIP1617 -> DIP1623. In a separate genome analysis, the region from DIP1645 to DIP1663 was considered as a pathogenicity island |
| GI_8 | 1,768,327 -1,781,812 bp | DIP1728 -> DIP1738 (10) | The IVOM score in this region is 18.08, and there are no highly expressed genes found in this region. |
| GI_9 | 1,861,303 -1,900,281 bp | DIP1811 -> DIP1857 (46) | This region contains one putative phage integrase, one putative phage terminase protein, and three tRNA genes. IslandViewer also predicted the genomic region from DIP1811 to DIP1841 to be a GI, and the region of DIP1817 -> DIP1837 was reported to a pathogenicity island in [17]. |
| GI_10 | 1,993,436 -2,020,291 bp | DIP1941 -> DIP1971 (31) | Four putative transposase genes, one Integrase gene and one tRNA gene are found in this predicted GI. The IVOM score in this region is 20.36. Islandviewer predicted two genomic Islands within this region, i.e., DIP1944 -> DIP1951, and DIP1960 -> DIP1964. |
| GI_11 | 2,043,495 -2,149,289 bp | DIP1993 -> DIP2098 (105) | This region contains three putative transposase genes including putative transposase for insertion element. The overall IVOM score is 20.18. IslandViewer predicted two GIs, DIP2015 -> DIP2022 and DIP2046 -> DIP2050. In another genome analysis study, two regions, DIP2010 -> DIP2015, and DIP2066 -> DIP2093, were considered to be pathogenicity islands. |
| GI_12 | 2,208,692 -2,237,426 bp | DIP2142 -> DIP2163 (21) | In this region, several GI related genes have been found, including putative bacteriophage holin protein, putative DNA-binding bacteriophage protein, and tRNA gene. In a separate genome study, researcher considered the region of DIP2148 -> DIP2168 to be a pathogenicity island. |
| GI_13 | 2,242,597 -2,275,566 bp | DIP2169 -> DIP2191 (22) | The IVOM score in this region is 15.42, which implies that this region contains different sequence composition from the rest of its host genome. |
| GI_14 | 2,284,782 -2,335,407 bp | DIP2199 -> DIP2245 (46) | The IVOM score in this region is 21.85. IslandViewer detected two genomic islands in this region, i.e., DIP2204 -> DIP2209, and 2) DIP2236 -> DIP2239. In addition, a separate genome analysis revealed that there was one pathogenicity island within this genomic region, which was from DIP2208 to DIP2234. |
Table 2: A list of fourteen predicted genomic islands in the genome of C. diphtheria NCTC13129.
In general, these fourteen genomic islands have higher IVOM scores, as shown in the second outer circle in Figure 8. Many of these islands also contain phage integrase, transposase and tRNA genes. In addition, most of our predicted genomic islands overlapped with those identified pathogenicity islands in previous studies [28]. For instance, the first predicted genomic island GI_1 covers 404 genes from gene DIP0021 to gene DIP0425. This genomic island contains two integrase genes, five phage genes, ten tRNA genes and five transposon related genes. The overall IVOM score in this region is 18.19, significantly higher than that of in non-GI region (typically <10).
We compared our predicted GI_1 with the predictions of IslandViewer [20], and we found that IslandViewer also considered this region as a GI region. In IslandViewer, however, this region was predicted as ten genomic islands, including: DIP0023 → DIP0035, DIP0057 → DIP0061, DIP0176 → DIP0179, DIP0219 → DIP0226, DIP0242 → DIP0249, DIP0281 → DIP0290, DIP0293 → DIP0294, DIP0330 → DIP0333, DIP0361 → DIP0364, and DIP0407 → DIP0418. In a separate genome analysis [28] has shown that this genomic region is pathogenic related. Four pathogenicity islands in this region were detected, including DIP0180 → DIP0222, DIP0223 → DIP0244, DIP0282 → DIP0287, and DIP0334 → DIP0357. All four pathogenicity islands are covered in our prediction result. Whether this genomic region is a single insertion event or separate insertion events needs to be further investigated. However, detailed studies on these predicted GIs by various algorithms make us to believe that smaller adjacent GIs, such as DIP0180 → DIP0222 and DIP0223 → DIP0244, might be a single GI.
Database of predicted genomic islands
We downloaded fully sequenced microbial genomes from the NCBI web server (ftp://ftp.ncbi.nih.gov/genomes/Bacteria), and ran our program GIHunter to predict genomic islands for fully sequenced genomes. All predicted genomic islands are available on our genomic islands database (http://www5.esu.edu/cpsc/bioinfo/dgi). By providing predicted genomic islands, we hope that microbiologists and medical biologists can better understand the mechanism of gene transfer in bacterial and archaeal genomes.
To provide microbiologists a better visualization on predicted genomic islands, we used Circos program [30] to generate genomic island images, which can represent the relative positions of predicted GIs in the genomes. In addition, we want to show the evidences of predicted GI regions by displaying all GI-associated feature values in the corresponding positions. To this end, we can display all feature values by different circular ideograms. By aligning different feature circles along with the GI circle, we can identify which feature values are important when used in predicting GIs. Figure 8 shows an example of predicted genomic islands in the genome of C. diphtheria NCTC13129, where each circle represents one GI- associated feature, with the order of features from inside to outside: 1) tRNA; 2) Phage; 3) Integrase; 4) Transposons; 5) HEG; 6) Intergenic Distance; 7) Density; and 8) IVOM. The most outer circle (9) represents the predicted GIs locations.

Figure 8:An example of predicted genomic islands in the genome of C. diphtheria NCTC13129. Each circle represents the feature values of tRNA (1), Phage (2), Integrase (3), Transposase (4), HEG (5), AvgID (6), Gene density (7), and IVOM (8). The most outer circle shows the predicted fourteen GI regions.
In this paper, we have reported the development of an ensemble of decision tree model for classifying GI/no-GI genes, based on eight features: IVOM, density, Highly Expressed Gene, tRNA, integrase, transposase, intergenic distance, and phage. We have developed our program GIHunter based on the trained model to predict GIs in any genome. The prediction accuracy of our approach has shown to be more accurate than that of other sequence composition approaches. The improved performance of our approach over previous ones is due to the incorporation of multiple genomic island related features in our model. For example, the feature of IVOM is effective in general, but the incorporation of gene information such as integrase, transposase, and highly expressed genes will make the prediction more accurate. We hope our program, GIHunter (http://www.esu.edu/cpsc/che_lab/software/GIHunter), can help accurately detecting genomic islands in future sequenced genomes.
Additionally, in order to make non computational scientists to access predicted genomic islands easily, we have predicted genomic islands for more than 2,000 bacterial and archaeal genomes, and provided visualized GI images (http://www5.esu.edu/cpsc/bioinfo/dgi). Therefore, microbiologists and medical biologists could investigate the gene transfer or pathogenicity mechanisms of their interested organisms by looking into our predicted genomic islands.
In our future work, we will create the database of sub-categorized genomic islands. Different genomic islands perform different functions. Some of them may contain phage related genes which may cause disease in some cases, while others contain degradation or resistance genes. Therefore, it will be meaningful and important to categorize genomic islands based on their functions inside the genomic islands. We propose to categorize them into six groups, including pathogenicity island, degradation island, resistance island, metabolism island, symbiotic island, and secretion genomic island. We strongly believe that providing such sub-groups of islands will be beneficial to researchers in the microbial communities.
The high-throughput sequencing technologies have generated huge amounts of draft genomes, and have left us to annotate. It will be very useful to integrate genomic island prediction tools into the current genome annotation tools. Recently, GI-POP [31], a comprehensive annotation Web server, has been developed for ongoing genome project analysis.
GI-POP contains a sequence assembling tool, functional annotation tools, and support vector machine (SVM)-based genomic island genomic profile scanning (GI-GPS) tool. The GI-GPS tool, however, is mainly based on genomic and codon signature (including: codon usage frequency, dinucleotide frequency, codon adaption index, and GC content). We believe our GI tool GIHunter, which incorporates multiple sources, will be more accurately characterizing genomic island information for the future draft genomes. Thus, we will build a similar genome annotation pipeline that incorporates our GIHunter into current annotation tools in our future work.
This research was partially supported by President Research Fund, Faculty Professional Development & Research (FDR) major grant, and FDR mini grant at East Stroudsburg University, USA.