Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2009) Volume 2, Issue 7
The genus Phoma, a common plant pathogen is taxonomically controversial. The conventional systems of classification of Phoma are functional but require considerable expertise to apply, which have resulted in a highly polyphyletic genus Phoma. The advent of molecular taxonomic techniques offered a solution for many problems, which were out of reach for classical taxonomic approaches. The method of construction of phylogenetic tree based on molecular data is widely used to determine evolutionary relationships. In the present study, we have selected 28S, 18S, and 5.8S with ITS region nucleotide sequences, actin gene sequences and beta tubulin gene sequences for the in silico analysis of the evolutionary relationship. The main objectives of the study were to assess the genetic variations and relatedness along with the investigation, identification, classification and evolutionary relationships among the eleven selected Phoma species. Confirmation of our results has been done by applying various statistical tests. The results have revealed that species have a number of discrete, highly divergent, genetic units. In contrast, some species have high sequence similarity and identity to each other, which are found as distinct classically. Our phylogenetic analysis has revealed that first speciation event is quickly followed by a second speciation event in one of the two resulting population of Phoma species.
Keywords: Phoma, Phylogenetic analysis, Evolutionary relationship, Molecular evolution, Statistical tests.
Phoma is a ubiquitous fungus that inhabits the soil and plants. It is a common plant pathogen but rarely cause infection in humans. The genus Phoma contains almost 2000 species all over the world (Boerema et al., 2004). Most of the strains isolated from human infections have not been identified to species level. Color of the colony, morphology of the conidia, existence and structure of chlamydospores help in differentiation of the species from each other (de et al., 2000).
Phoma is a taxonomically controversial genus and is not fully understood. It belongs to the order Pleosporales. It is unique form of pycnidiales, which occurs ubiquitously and has been reported from a wide variety of hosts particularly from plant and soil. It has also been recovered from aquatic and aerial environment (Rai and Rajak, 1983), marine environment (Sugano et al., 1991), entomopathogenic (Narendra and Rao, 1974) and has been found to cause disease in human beings (Shukla et al., 1984; Baker et al., 1987; Rai, 1993).
The identification of Phoma species has been performed mainly on the basis of hosts, morphological and cultural criteria and were extensively studied by Boerema and his colleagues (1970). This method is widely used by researchers. However, the process is complicated, tedious and requires skilled manpower.
Now-a-days, the development of molecular taxonomic tools for the identification and differentiation is the basis for defining genera of phomoid fungi. Identification of Phoma and Phoma-like fungi could be greatly facilitated by a largely DNA-based system for classification.
Over the last few years, a database of rDNA sequences, actin gene sequences and beta tubulin sequences has been developed for Phoma species and published in NCBI (http://www.ncbi.nlm.nih.gov/ ). To evaluate the classification and identification of differentPhoma species, we conducted a phylogenetic study of Phoma species using partial sequence of 18S ribosomal RNA gene, 28S ribosomal RNA gene, complete sequence of 5.8S ribosomal RNA gene and internal transcribed spacer region sequences.
The 28S, 18S, and 5.8S molecules are formed by the processing of a single primary transcript from a cluster of identical copies of single gene, it is suitable to select these sequences for evolutionary relationships analysis. The rRNA is the most conserved (least variable) gene in all cells. For this reason, genes that encode the rRNA (rDNA) have been sequenced to identify an organism’s group, calculate related groups, and estimate rates of species divergence (Wuyts et al., 2002). The recovery and the analysis of rRNA genes directly from the environmental DNA provides a measure of investigating microbial populations of any habitat, eliminating dependance on isolation of pure cultures (Ward et al., 1990; Amann et al., 1995).
Actin is a globular and highly conserved protein found in all eukaryotic cells (the only known exception being nematode sperm) where it may be present at concentrations of over 100 µM. It is also highly-conserved protein, differing by no more than 20% in species as diverse as algae and humans. The typical actin gene has an approximately 100- nucleotide 5' UTR, a 1200-nucleotide translated region, and a 200-nucleotide 3' UTR. The majority of actin genes are interrupted by introns, with up to 6 introns in any of 19 wellcharacterised locations. The highly conserve nature of the family makes actin the favoured model for comparing the introns-early and introns-late models of intron evolution (Doherty and McMohan, 2008).
Tubulin is one of the members of a family of globular proteins. The most common members of the tubulin family are α-tubulin and β-tubulin, the proteins that make up microtubules. Microtubules are assembled from α- and β-tubulin. The antifungal drug Griseofulvin targets mictotubule assembly and has applications in cancer treatment (Howard and Hyman, 2003).
Small genetic veriations along with time may provide us the exact and most accurate information to reconstruct the evolutionary relationship among the Phoma species.
The advent of molecular taxonomic techniques offered a solution for many problems, which were not available for classical taxonomic approaches. Currently, the methods of construction of phylogenetic tree based on molecular data are widely used not only in systematic and comparative biology, but also in ecology, sociobiology and epidemiology (Hampl et al., 2001).
Phylogenetic analysis gives insight into how a family of related sequences has been derived during evolution. The evolutioinary relationships among the sequences are drawn as branches of tree (Figure 4, Figure 5 and Figure 6). The length and nesting of these branches reflects the degree of similarity between any two given sequences and the degree of dissimilarity between the genes represented by the nodes. The degree of dissimilarity is calculated when the sequences are compared. Sequences that are the most closely related are drawn as neighbouring branches on a tree. The conserved region analysis has also been performed as a basic need for the present computation.
Disparity Index Analysis
A common assumption in comparative sequence analysis is that the sequences have evolved with the same pattern of nucleotide substitution (homogeneity of the evolutionary process). Violation of this assumption is known to adversely impact the accuracy of phylogenetic inference and tests of evolutionary hypothesis. A disparity index (ID), measures the observed difference in evolutionary patterns for a pair of sequences. On the basis of this index, Kumar and Gadagkar (2001) have developed a Monte Carlo procedure to test the homogeneity of the observed patterns. This test does not require a prior knowledge of the pattern of substitutions, extent of rate heterogeneity among sites, or the evolutionary relationship among sequences. Computer simulations have shown that the ID-test is more powerful than the commonly used X2 c -test under a variety of biologically realistic models of sequence evolution (Kumar and Gadagkar, 2001; Kumar et al., 2001). Thus, the proposed test can be used as a diagnostic tool to identify genes and lineages that have evolved with substantially different evolutionary processes as reflected in the observed patterns of change. Identification of such genes and lineages is an important early step in comparative genomics and molecular phylogenetic studies to discover evolutionary processes that have shaped the genomes of the organisms.
Test of Neutrality
DNA polymorphisms are powerful sources of information for studying the evolution of a population. Whether a locus or region from which a DNA sample has been taken evolves neutrally or under natural selection is of considerable interest in evolutionary study and can be examined using a statistical test designed for DNA polymorphisms (Yunxin, 1996; Goto et al., 1999). A popular statistical test proposed by Tajima,(1989) is:
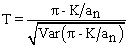
where, π is the mean number of nucleotide differences between two sequences, K is the number of segregating sites, n is sample size and
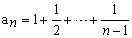
An essential parameter in the theory of neutral evolution is θ = 4Nµ, where N is the effective population size and µ is the mutation rate per sequence per generation. Almost all summary statistics of DNA polymorphisms are related to this parameter.
We have selected Phoma species whose sequences of rDNA, actin gene (act1) and beta tubulin are present. After searching the GenBank, we found sequences of only 11 species of Phoma with the above mentioned gene records. Thus, we restricted our analysis of Phylogenetic inference around these 11 selected Phoma species.
The major objectives of the study were to assess the genetic variations and genetic relatedness along with the investigation of identification, classification and evolutionary relationships among the eleven selected Phoma species namely P. exigua Desmazières, P. medicaginis Malbr. and Roum, two isolates of P. pinodella Morgan-Jones Burch,P. betae Frank, Phoma sp. WAC 4738, Phoma sp. WAC 4736, Phoma sp. WAC 4741, Phoma sp. WAC 7980, Phomaeupyrena Sacc., and Phoma sp. OMT 5, by using rDNA sequences, actin gene (act1) sequences and beta tubulin gene sequences.
The data used in reconstruction of a DNA-based phylogenetic tree are obtained by comparing nucleotide sequences and performing multiple sequence alignment to find the sequence similarity among the selected 11 sequences. This is the critical part of the entire analysis because if the alignment is incorrect then the resulting tree will definitely not be true tree. Multiple sequence alighnments yield information into the evolutionary history of the sequences that are most similar and likely to be derived from a common ancestor sequence.
Making the biological information available for analysis and developing applications is an important task. We used such previously available and published information from NCBI (http://www.ncbi.nlm.nih.gov/). There are large number of databases in the public and private domain. rDNA, actin gene (act1) and beta tubulin sequences of a nucleotide encoded by different species may or may not be similar
Sequence Retrieval and Analysis
To compare the similarity or diversity, the nucleotide sequences need to be downloaded. 18S ribosomal RNA gene, 28S ribosomal RNA gene, complete sequence of 5.8S ribosomal RNA gene and internal transcribed spacer region sequences of 11 selected Phoma species namely P. exigua (EU555533), P. medicaginis (AY831563), P. pinodella strain CBS 318.90 (EU573028), P. pinodella strain WAC 7978 (AY831556), P. betae (EU594572), Phoma sp. WAC 4738 (AY831560), Phoma sp. WAC 4736 (AY831561), Phoma sp. WAC 4741 (AY831559), Phoma sp. WAC 7980 (AY831555), Phoma eupyrena (EU573014) and Phoma sp. OMT 5 (AY831554) were retrieved from NCBI: GenBank. (http://www.ncbi.nlm.nih.gov/).
The actin gene sequences of P. exigua (AY831521), P. medicaginis (AY831530), P. pinodella strain CBS 318.90 (AY831529), P. pinodella strain WAC 7978 (AY831523), P. betae (AY748973), Phoma sp. WAC 4738 (AY831527), Phoma sp. WAC 4736 (AY831528), Phoma sp. WAC 4741 (AY831526), Phoma sp. WAC 7980 (AY831522), Phoma eupyrena (AY748975) and Phoma sp. OMT 5 (AY831519) were also retrieved.
Moreover, beta tubulin gene sequences of P. exigua (AY831509), P. medicaginis (AY831518), P. pinodella strain CBS 318.90 (AY831517), P. pinodella strain WAC 7978 (AY831511), P. betae (AY749021), Phoma sp. WAC 4738 (AY831515), Phoma sp. WAC 4736 (AY831516), Phoma sp. WAC 4741 (AY831514), Phoma sp. WAC 7980 (AY831510), Phoma eupyrena (EU541415) and Phoma sp. OMT 5 (AY831507) have been retrieved from the same resource mentioned above. The alignment between three or more sequences is the multiple sequence alignment. The alignment of nucleotide sequences can reveal whether any evolutionary relationship exist between the sequences.
Multiple Sequence Alignment
The multiple sequence alignment was performed using online software CLUSTAL W (Thompson et al., 1994) version 2.2.5 (http://www.ebi.ac.uk/Tools/clustalw2/index.html). The pair wise distance and standard error calculations were analyzed including transition (Ts) and transversion (Tv) substitutions (weight = 0.5) through the Kimura 2-parameter model (Kimura, 1980).
The conserved regions (Table 1) have been searched by using BioEdit version 5.0.6 (Tom Hall, 2001) with the minimum segment length of 15 nucleotides per sequence and maximum average entropy of 0.2. Maximum entropy per position was 0.2 with gaps limited to 2 per segment. Contiguous gaps limited to 1 in any segment.
Molecular Evolutionary Relationships Analysis
The evolutionary history was inferred using the Maximum Parsimony method (MP) (Eck and Dayhoff, 1966). The bootstrap consensus trees inferred from 1000 replicates were taken to represent the evolutionary history of the taxa analyzed. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) was shown next to the branches (Felsenstein, 1985). The MP trees were obtained using the Close-Neighbor-Interchange algorithm (Nei and Kumar, 2000) with search level 3 in which the initial trees were obtained with the random addition of sequences (10 replicates). The trees were drawn to scale; with branch lengths calculated using the average pathway method (Nei and Kumar, 2000; Kumar et al., 2004) and were in the units of the number of changes over the whole sequence. All positions containing gaps and missing data were eliminated from the dataset (Complete Deletion option). In all there were 501, 396 and 300 positions in rDNA, actin gene and beta tubulin gene sequences respectively in the final dataset, out of which 21 for rDNA, 37 for actin gene and 34 for beta tubulin genes were parsimony informative. Phylogenetic analyses were conducted in MEGA4 (Tamura et al., 2007).
Nucleotide Substitution Analysis
The rate of nucleotide substitution (r) was allowed to vary from branch to branch, so that it would be convenient to measure evolutionary time in terms of the expected number of substitutions (v=rt). The pattern of nucleotide substitution was computed by using (HKY) model (Hasegawa et al., 1985b). The nucleotide substitutions matrices obtained from different branches were averaged by weighting each matrix and the number of inferred substitutions for the branch. Further averages of matrices for two different genes were also calculated by the same weighting method. Table 4 shows the relative frequencies of the twelve different nucleotide substitutions for three different Phoma sequences (rDNA, actin (act1) gene and beta tubulin gene).
Estimation of Net Base Composition Bias Disparity between Sequences
Disparity Index (ID) per site was calculated for all sequence pairs. Values greater than 0 indicate that the larger differences in base composition bias than expected, based on evolutionary divergence between sequences and by chance alone. Codon positions included were 1st+2nd+3rd+Noncoding for act1 and beta tubulin genes (Table 5, 6 and 7). All the positions containing gaps and missing data were eliminated from the dataset (Complete deletion option).
Neutrality Analysis
The Tajima test statistic (Tajima, 1989) was estimated using MEGA4. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option). The abbreviations used are as follows: m = number of sites, S = Number of segregating sites, ps = S/m, Θ = ps/ a1, and π = nucleotide diversity. D is the Tajima test statistic (Tajima, 1993).
Conserved Region Search
After multiple sequence alignment analysis, we obtained 5 conserved regions in rDNA, 7 in actin gene (act1) with a minimum length of 15 nucleotides and maximum entropy 0.2. No conserved region in beta tubulin gene was found. Conserved regions were generated by BioEdit version 7.0.9 (Hall, 1999).
To confirm these findings, we used an entropy plot analysis of the alignment (Figure 1, 2 and 3). rDNA undergo nucleotide substitutions from 1 to 47, then remain conserved after this period up to 77 (data not shown). From 254, the conserved regions would again start up to 606 with some nucleotide substitution regions. Similarly, actin gene (act1) is conserved after 300 up to 602, with some exceptions of nucleotide substitutions (data not shown). Alignment of beta tubulin gene sequences could not show any significant conserved region.

Figure 1: Entropy plot of rDNA sequence alignment of 11 species of Phoma.
Entropy analysis of rDNA: Participated nucleotides: from nucleotide position 1 to 635. X- axis = alignment position; Y- axis = entropy.

Figure 2: Entropy plot of actin gene (act1) sequence alignment of 11 species of Phoma.
Entropy analysis of actin gene: Participated nucleotides: from nucleotide position 1 to 903. X- axis = alignment position; Y- axis = entropy.

Figure 3: Entropy plot of beta tubulin sequence alignment of 11 species of Phoma.
Entropy analysis of beta tubulin gene: Participated nucleotides: from nucleotide position 1 to 1279. X- axis = alignment position; Y- axis = entropy.
| Gene (sequence) |
Conserved regions found | Position | Sequence | Segment length |
| rDNA | 5 | 55 to 78 | TGAACCTGCGGAAGGATCATTACC | 24 |
| 254 to 425 | TAATAGTTACAACTTTCAACAACGGATCTCTTGGTT CTGGCATCGATGAAGAACGCAGCGAAATGCGATAA GTAGTGTGAATTGCAGAATTCAGTGAATCATCGAAT CTTTGAACGCACATTGCGCCCCTTGGTATT CCATGG GGCATGCCTGTTCGAGCGTCAT TTGTACC |
172 | ||
| 435 to 458 | TGCTTGGTGTTGGGTGTTTGTCTC | 24 | ||
| 482 to 500 | AAAACAATTGGCAGCCGGC | 19 | ||
| 573 to 606 | CACTCTTGACCTCGGATCAGGTAGGGATACCCGC | 34 | ||
| Actin (act1) | 7 | 300 to 314 | CTACCTCATGAAGAT | 15 |
| 369 to 386 | TGACATCAAGGAGAAGCT | 18 | ||
| 409 to 425 | GAGCAGGAGATCCAGAC | 17 | ||
| 472 to 500 | GGTCAGGTCATCACCATTGGCAACGAGCG | 29 | ||
| 541 to 565 | GGTCTTGAGAGCGGTGGTATCCACG | 25 | ||
| 574 to 602 | TTCAACTCCATCATGAAGTGCGATGTCGA | 29 | ||
| 689 to 711 | AGTCTGGTGGTACCACCATGTAC | 23 | ||
| Beta tubulin | 0 | - | -------------- | - |
Minimum segment length (actual for each sequence): 15, Maximum average entropy: 0.2, Maximum entropy per position: 0.2, Gaps limited to 2 per segment, Contiguous gaps limited to 1 in any segment.
Table 1: Conserved regions found in the rDNA, Actin gene (act1) and beta tubulin gene sequences of 11 Phoma species.
Phylogenetic Inference
Figure 4, Figure 5 and Figure 6 show the phylogenetic relationships among eleven selected Phoma species.

Figure 4: Evolutionary relationships of 11 taxa of Phoma on the basis of rDNA sequence alignment.
A phylogram of 11 selected Phoma species on the basis of rDNA sequence alignment that indicates the relationship between the species and also conveys a sense of time or rate of evolution. The branching pattern, which is rooted by using Phoma betae as the out-group, was generated by the neighbor-joining method. Bootstrap values (n = 1000 replicates) are given for each node having 50% or greater support. There were a total of 501 positions in the final dataset, out of which 21 were parsimony informative.

Figure 5: Evolutionary relationships of 11 taxa of Phoma on the basis of Actin gene (act1) sequence alignment.
A phylogram of 11 selected Phoma species on the basis of actin (act1) gene sequence alignment that indicates the relationship between the species and also conveys a sense of time or rate of evolution. The branching pattern, was generated by the neighbor-joining method. Bootstrap values (n = 1000 replicates) are given for each node having 50% or greater support. There were a total of 396 positions in the final dataset, out of which 37 were parsimony informative.

Figure 6: Evolutionary relationships of 11 taxa of Phoma on the basis of beta tubulin gene sequence alignment.
A phylogram of 11 selected Phoma species on the basis of beta tubulin sequence alignment that indicates the relationship between the species and also conveys a sense of time or rate of evolution. The branching pattern, which is rooted by usingPhoma betae as the out-group, was generated by the neighbor-joining method. Bootstrap values (n = 1000 replicates) are given for each node having 50% or greater support. There were a total of 300 positions in the final dataset, out of which 34 were parsimony informative.
| rDNA | A | T | C | G |
|---|---|---|---|---|
| A | - | 4.85 | 4.13 | 20.58 |
| T | 3.98 | - | 11.46 | 3.96 |
| C | 3.98 | 13.46 | - | 3.96 |
| G | 20.67 | 4.85 | 4.13 | - |
| Actin | A | T | C | G |
| A | - | 4.28 | 5.6 | 8.14 |
| T | 3.88 | - | 27.42 | 4.44 |
| C | 3.88 | 20.95 | - | 4.44 |
| G | 7.11 | 4.28 | 5.6 | - |
| Beta tubulin | A | T | C | G |
| A | - | 2.24 | 2.81 | 18.49 |
| T | 1.84 | - | 27.42 | 2.56 |
| C | 1.84 | 21.86 | - | 2.56 |
| G | 13.3 | 2.24 | 2.81 | - |
Table 2: Maximum Composite Likelihood Estimate of the Pattern of Nucleotide Substitution in rDNA, Actin (act1) gene and beta tubulin gene sequences of Phoma species.
18S ribosomal RNA gene, 28S ribosomal RNA gene, complete sequence of 5.8S ribosomal RNA gene and internal transcribed spacer region sequences are comparatively analyzed with actin and beta tubulin gene sequences of the same species. Phylogenetic analysis using bootstrap values (n = 1000 replicates) are given for each node having 50% or greater support revealed that some of the sequences were>95 % similar to each other e.g., P. pinodella strain CBS 318.90: P. pinodella strain WAC 7978 : P. eupyrena and Phoma sp. WAC 4738 : Phoma sp. WAC 4736 : Phoma sp. WAC 4741 : Phoma sp. WAC 7980 : Phoma sp. OMT 5. This indicates that these species are evolved at the same time with orthologs due to a speciation event. Isolation of these species may lead to successive lineages.
Figure 4, 5 and 6 indicated that P. betae which is rooted, as an out-group is the most distinct species among all the eleven species of Phoma. Figure 5 expresses the Phoma sp. WAC 4738, Phoma sp. WAC 4736, Phoma sp. WAC 4741, Phoma species WAC 7980 and Phoma sp. OMT 5 comes in a cluster. These species are closer to P. medicaginis than the rest of the Phoma species.
Maximum Parsimony method was used for phylogenetic inference. To evaluate the accuracy and reliability of our phylogenetic tree, statistical tests (described below) were applied to the topologies. The reliability of branch length estimates was tested by bootstrap method. Felsenstein’s (1985), bootstrap test is one of the most commonly used tests of the reliability of an inferred tree.
| rDNA | Transition (Ts) | Transversion (Tv) | ||
| Pattern | entries | Pattern | entries | |
| A – T | 4.85 | A – C | 4.13 | |
| T – A | 3.98 | C – A | 3.98 | |
| C – G | 3.96 | T – G | 3.96 | |
| G – C | 4.13 | G – A | 4.85 | |
| Actin | Transition (Ts) | Transversion (Tv) | ||
| Pattern | entries | Pattern | entries | |
| A – T | 4.28 | A – C | 5.60 | |
| T – A | 3.88 | C – A | 3.88 | |
| C – G | 4.44 | T – G | 4.44 | |
| G – C | 5.60 | G – A | 4.28 | |
| Beta tubulin | Transition (Ts) | Transversion (Tv) | ||
| Pattern | entries | Pattern | entries | |
| A – T | 2.24 | A – C | 2.81 | |
| T – A | 1.84 | C – A | 1.84 | |
| C – G | 2.56 | T – G | 2.56 | |
| G – C | 2.81 | G – A | 2.24 | |
Table 3: Rates of different transitional and transversional substitutions in the rDNA, Actin gene (act1) and beta tubulin gene sequences of 11 Phoma species.
| A | T | C | G | K1 | K2 | Ts/Tv bias | |
|---|---|---|---|---|---|---|---|
| rDNA | 0.235 | 0.287 | 0.244 | 0.234 | 5.197 | 2.775 | 1.997 |
| Actin (act1) | 0.213 | 0.235 | 0.308 | 0.244 | 1.834 | 4.901 | 1.999 |
| Beta tubulin | 0.195 | 0.237 | 0.297 | 0.271 | 7.212 | 9.744 | 5.064 |
The overall transition/transversion bias for these 3 sequence types have been calculated and analyzed by using the formula R = [A*G*k1 + T*C*k2] / [(A+G)*(T+C)]. Where, k1 is the transition/transversion rate ratio for purine and k2 is the transition/transversion rate ratio for pyrimidine. All positions containing gaps and missing data were eliminated from the dataset (Complete-deletion option).
Table 4: The nucleotide frequencies, the transition/transversion rate ratios and the overall transition/transversion bias in the rDNA, Actin gene (act1) and beta tubulin gene sequences of 11 Phoma species.
Nucleotide Substitution Analysis
Table 2 shows the Maximum Composite Likelihood estimate of the pattern of Nucleotide Substitution in rDNA, actin (act1) gene and beta tubulin gene sequences of eleven selected Phoma species. Each entry shows the probability of substitution from one base (row) to another base (column) instantaneously. Only entries within a row have been compared. Rates of different transitional substitutions are shown in bold and those of transversional substitutions are shown in italics. The pattern of nucleotide substitution (Table 3) implied that A—T transitions in rDNA> was greater than rest of the substitution patterns (T—A, C—G and G—C). Conversely, in the actin and beta tubulin sequence analysis, G—C transition was greater than the rest of the substitution. Moreover, rDNA nucleotide composition analysis has revealed that most of the rDNA sequences are A+T rich, whereas actin gene and beta tubulin gene sequences are G+C rich. These observations code that the transitions are dependent upon the percentage of G + C or A+T content within the sequences. The analysis of transversions revealed that, in the rDNA, G—A is greater and in actin and beta tubulin, A—C is greater than the rest of the substitution patterns.
The nucleotide frequency comparison (Figure 7, 8, 9 and 10) (Table 4) revealed that, in rDNA A and T were frequent that G and C, whereas; G and C nucleotide frequency were greater in actin and beta tubulin. This will support the symptoms that the rDNA has greater A—T transition. rDNA consist of more number of purines while, actin and beta tubulin have more number of pyrimidines.

Figure 7: Comparison of nucleotide substitution patterns in rDNA sequences of 11 taxa of Phoma.

Figure 8: Comparison of nucleotide substitution patterns in Actin (act1) gene sequences of 11 taxa of Phoma.

Figure 9: Comparison of nucleotide substitution patterns in Beta tubulin gene sequences of 11 taxa of Phoma.

Figure 10: Comparison of nucleotide frequencies in rDNA, Actin gene and Beta tubulin gene sequences of 11 taxa of Phoma.
Among the four different types of transitional changes, the G—A change is most frequent to all positions. In the third position the changes between T and C are as frequent as the changes between A—G. Among the transversional changes, the C—A change is always less frequent than the A—C change. Also, G—T changes are more frequent than T—G in rDNA nucleotide pattern substitution, whereas T— G is more frequent in act-1 and beta tubulin genes analysis. Among the transversional changes G—T and T—G changes are often most frequent.
The nucleotide frequencies for rDNA sequences were 0.235 (A), 0.287 (T), 0.244 (C), and 0.234 (G). For actin gene, the nucleotide frequencies were 0.213 (A), 0.235 (T), 0.308 (C), and 0.244 (G). The nucleotide frequencies for beta tubulin genes were 0.195 (A), 0.237 (T), 0.297 (C), and 0.271 (G). The transition/transversion rate ratios for rDNA, actin gene and beta tubulin gene sequences were k1 = 5.197 (purines) and k2 = 2.775 (pyrimidines), k1 = 1.834 (purines) and k2 = 4.901 (pyrimidines) and k1 = 7.212 (purines) and k2 = 9.744 (pyrimidines) respectively. The overall transition/transversion bias for these 3 sequence types are; R = 1.997, R = 1.999 and R = 5.064 respectively; where R = [A*G*k1 + T*C*k2] / [(A+G)*(T+C)]. The missing data and the positions containing gaps were removed by complete deletion. All calculations were conducted in MEGA4. These outputs reveal that multiple substitutions had occurred at several sites.
Additionally, the relative rate of phylogeny has been calculated by Tajima’s relative rate test (Tajima, 1993) under molecular clock hypothesis, E(nijk) = E(njik) irrespective of the substitution model and whether or not the substitution rate varies with the site. If this hypothesis is rejected, then the molecular clock hypothesis can be rejected for this set of sequences (Nei and Kumar, 2000).
These calculations confirm that the evolutionary rate lineages among all the species are unsystematic. It also confirms that P. betae is highly distinct species among all the eleven species of Phoma.
Disparity Index Analysis
Disparity Index (ID) per site was calculated for all sequence pairs (Table 5, 6 and 7). There were a total of 501, 396 and 300 positions respectively in the final dataset.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | |||||||||||
| 2 | 0.000 | ||||||||||
| 3 | 0.000 | 0.000 | |||||||||
| 4 | 0.000 | 0.000 | 0.000 | ||||||||
| 5 | 0.000 | 0.000 | 0.000 | 0.000 | |||||||
| 6 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | ||||||
| 7 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |||||
| 8 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | ||||
| 9 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |||
| 10 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | ||
| 11 | 0.000 | 0.014 | 0.054 | 0.000 | 0.036 | 0.036 | 0.036 | 0.036 | 0.036 | 0.016 |
No. of Taxa: 11, No. of Sites: 501. The species arrangement is as follows:
(1) P. pinodella sp. CBS318.90 (2) P. pinodella sp. WAC7978 (3) P. exigua (4) P. eupyrena (5) P. sp. WAC4738 (6) P. sp. WAC4736 (7) P. sp. WAC4741 (8) P. sp. WAC7980 (9) P. sp. OMT 5 (10) P. medicaginis (11) P. betae. Gaps/Missing Data: Complete Deletion
Table 5: Estimates of net base composition bias disparity between sequences.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | |||||||||||
| 2 | 0.000 | ||||||||||
| 3 | 0.000 | 0.000 | |||||||||
| 4 | 0.000 | 0.000 | 0.000 | ||||||||
| 5 | 0.003 | 0.003 | 0.003 | 0.003 | |||||||
| 6 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | ||||||
| 7 | 0.020 | 0.020 | 0.020 | 0.020 | 0.003 | 0.000 | |||||
| 8 | 0.126 | 0.126 | 0.126 | 0.126 | 0.086 | 0.073 | 0.000 | ||||
| 9 | 0.515 | 0.515 | 0.515 | 0.515 | 0.396 | 0.419 | 0.321 | 0.164 | |||
| 10 | 0.515 | 0.515 | 0.515 | 0.515 | 0.396 | 0.419 | 0.321 | 0.164 | 0.000 | ||
| 11 | 0.328 | 0.328 | 0.328 | 0.328 | 0.230 | 0.263 | 0.240 | 0.146 | 0.000 | 0.000 |
No. of Taxa: 11, Codon Positions: 1st+2nd+3rd+Noncoding, No. of Sites: 396. The species arrangement is as follows:
(1) P. sp. WAC4738 (2) P. sp. WAC4736 (3) P. sp. WAC4741 (4) P. sp. WAC7980 (5) P. sp. OMT 5 (6) P. medicaginis (7) P. eupyrena (8) P. betae (9) P. pinodella sp. CBS318.90 (10) P. pinodella sp. WAC7978 (11) P. exigua
Table 6: Estimates of net base composition bias disparity between sequences.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | |||||||||||
| 2 | 0.000 | ||||||||||
| 3 | 0.000 | 0.000 | |||||||||
| 4 | 0.000 | 0.000 | 0.000 | ||||||||
| 5 | 0.000 | 0.000 | 0.000 | 0.000 | |||||||
| 6 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | ||||||
| 7 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |||||
| 8 | 0.000 | 0.000 | 0.000 | 0.003 | 0.003 | 0.003 | 0.003 | ||||
| 9 | 0.000 | 0.000 | 0.000 | 0.007 | 0.007 | 0.007 | 0.007 | 0.007 | |||
| 10 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.003 | 0.017 | ||
| 11 | 3.060 | 3.060 | 2.973 | 2.553 | 2.553 | 2.553 | 2.553 | 2.467 | 2.420 | 3.470 |
No. of Taxa: 11, Codon Positions: 1st+2nd+3rd+Noncoding, No. of Sites: 300. The species arrangement is as follows:
1) P. pinodella sp. CBS318.90 (2) P. pinodella sp. WAC7978 (3) P. eupyrena (4) P. sp. WAC4738 (5) P. sp. WAC4736 (6) P. sp. WAC4741 (7) P. sp. WAC7980 (8) P. sp. OMT 5 (9) P. medicaginis (10) P. exigua (11) P. betae
Table 7: Estimates of net base composition bias disparity between sequences.
The disparity index analysis of three types of gene sequences of Phoma is comparable. Actin gene has greater range of disparity than rDNA and beta tubulin. Order of this disparity is rDNA < beta tubulin < actin (act1). The kin observation shows the scope for division of all these species in two groups. One includes Phoma species WAC4738,Phoma species WAC 4736, Phoma species WAC 47341,Phoma species WAC 7980 and Phoma species OMT-5 and P. medicaginis. The nearest neighbour of this cluster is P. eupyrena. The next group includes rest of the Phoma species, except the out group P. betae.
Neutrality Analysis
The relationship between the two estimates of genetic variation at the DNA level, namely the number of segregating sites and the average number of nucleotide differences estimated from pairwise comparison, was investigated. Statistical method for testing the neutral mutation hypothesis was developed. This method needs only the data of DNA polymorphism, namely the genetic variation within population at the DNA level. In our analysis, beta tubulin gene has the highest number of segregation size (Table 8). Therefore, it has maximum diversity than rDNA sequence and actin gene. The order of nucleotide diversity is rDNA < actin < beta tubulin.
| m | S | ps | Θ | π | D |
|---|---|---|---|---|---|
| 11 | 71 | 0.141717 | 0.048384 | 0.039448 | -0.879743 |
| 11 | 62 | 0.156566 | 0.053454 | 0.056657 | 0.284596 |
| 11 | 130 | 0.433333 | 0.147947 | 0.119394 | -0.928064 |
The Tajima test statistic was estimated. All positions containing gaps and missing data were eliminated from the dataset (Complete deletion option). The abbreviations used are as follows: m = number of sites (no. of sequences), S = Number of segregating sites, ps = S/m, Θ = ps/a1, and π = nucleotide diversity. D is the Tajima test statistic (Nei and kumar, 2000).
Table 8: Results from Tajima’s Neutrality Test for the rDNA, Actin gene (act1) and beta tubulin gene sequences of 11 Phoma species.
Rajak and Rai (1993) reported that the growth and color of the colony helps in differentiating the species in the genusPhoma. Slow growth of P. fimeti under various physiological conditions clearly differentiate it from other species. Production of pigments and metabolites in the mycelium as well as in the medium has often been found to be useful differentiating character in certain species of Phoma. It is an oxidation reaction of metabolite “E” (Rajak and Rai, 1983). Although pigment production is slightly variable character it may still have importance as a taxonomic character when combined with other characters (Rai, 1981).
Shape and size of pycnidia have been considered as important taxonomic character in many genera of sphaeropsidales including Phoma. According to Singh, (1974) shape and size of pycnidia in P. exigua were highly variable characters under different conditions.
The conventional systems of classification of Phoma (Boerema, 1997, 1973; Rai, 1981; Boerema et al., 2004) are systems that are functional but require considerable expertise to apply.
A little work has been done on biochemical aspect of taxonomic criterion for species differentiation in Phoma. Rai, (1985) tried cholesterol as a taxonomic marker for the differentiation of species within the genus Phoma. They reported significant similarities even among morphologically different species and vice versa. They developed a key to the identification of Phoma species based on the morphological and cultural characters.
If host alone is taken to separate the taxa, it may result in considerable confusion if the identification based on host itself proves to be wrong or where a morphological species might show different characters with changed environmental conditions such as pigment production on agar medium. To overcome the conventional method, it was thought to study genetic diversity or relatedness using molecular markers, in order to create a more realistic and usable classification of Phoma and related fungi.
Variations within the internal transcribed spacer (ITS-1, 5.8 S gene and ITS-2) region of the DNA to characterize the phylogenetic relationships among Phoma ligulicola isolates infecting pyrethrum crops in Tasmania was studied (Pethybridge et al., 2004). Edgcomb and his group in 2001 demonstrated the tubulin gene phylogeny to elucidate relationships among “jakobids” and other early-diverging eukaryotic lineages. They found that tubulin gene phylogenies were in general agreement with mitochondrial gene phylogenies and ultra-structural data indicated that the “jakobids” may be polyphyletic.
Phylogenetic analysis plays an important role in the investigation of species diversity as well as novel species identification (Surakasi et al., 2007). Redecker et al. (1999) found that phylogenetic analysis of a dataset of fungal 5.8S rDNA sequences shows highly divergent copies of internal transcribed spacers reported from Scutellospora castanea of Ascomycete origin.
The rDNA based phylogenetic analysis and culture dependent phenotypic characterization of the cultivable bacterial diversity of alkaline Lonar Lake in India has been carried out (Joshi et al., 2007). They found that few bacteria were found in high G+C group. These isolates were associated with different phylum belonging to different families.
Arenal et al. (1999) performed the phylogenetic relationship between Epicoccum nigrum and Phoma epicoccina which was assessed by means of sequencing of the ITS regions of rDNA. The analysis of the sequences from five isolates of E. nigrum and four of P. epicoccina suggests that both entities represent the same biological species.
The assumption is that the gene tree, based on molecular data with all its advantages, will be a more accurate and less ambiguous representation of the species tree than that obtained by morphological comparisons. This assumption is often correct, but it does not mean that the gene tree is the same as the species tree (Brown, 2002). Drouin et al. (1995) used Giardia lamblia actin sequence to root the phylogenetic trees based on 65 actin protein sequences from 43 species. The tree was congruent with small-subunit rRNA trees in that it showed that oomycetes were not related to higher fungi; that kinetoplatid protozoans, green plants, fungi and animals were monophyletic groups; and that the animal and fungal lineages share a more recent common ancestor than either does with the plant lineage. Moreover, Matheucci, (1995) had suggested that the actin gene (act) can be used as a selectable marker for the use as a homologous promoter to direct expression of hygromycin-B-resistanceencoding gene.
The findings of Keeling et al., (2000), had coded that ever since the first phylogenetic evidence from tubulins that microsporidia are related to fungi. The resulting beta tubulin phylogeny was supposed to be in general agreement with what is believed to be the organism phylogeny of the two groups and had shown that the microsporidian beta tubulins emerge from within the fungal clade. These results provided the first clear demonstration that microsporidia evolved from a fungus. Kumar and Gadagkar, (2001) encoded an application of the disparity index (ID) test in an analysis of 3789 pairs of orthologous human and mouse protein-coding genes revealed that the observed evolutionary patterns in neutral sites are not homogeneous in 41% of the genes, apparently due to shifts in G + C content.
Our results suggest that the phylogenetic analysis reveals a rapid analysis of evolutionary relationship, which can help to identify and classify the species in different groups of Phoma. It also helps in the study of species divergence during speciation events. It also infers that Phoma betae is most distinct species among all.
Boerema et al., (1965) commented that P. pinodella is nothing but a variety of P. medicaginis. But we found that these two species had come in two different clusters. On the basis of our findings we propose that these two species are different. Indeed, Phoma sp. WAC 4738, Phoma sp. WAC 4736, Phoma sp. WAC 4741, Phoma sp. WAC 7980 and Phoma sp. OMT 5 could form a cluster with P. medicaginis. However, we found that P. pinodella and P. eupyrena could come within a cluster.
In conclusion, it is important to note despite the selection of taxonomically well resolved taxa, the results have revealed a number of species e.g. P betae and P. exigua, have a number of discrete, highly divergent, genetic units as compared to other selected Phoma species. In contrast, some species have high sequence similarity and identity to each other. The degree of divergence among these species reflects the species that have long period of isolation. The results are comparable with classical systematics, which codes the diversity of P. medicaginis and Phoma sp. WAC 4738, Phoma sp. WAC 4736, Phoma sp. WAC 4741, Phoma sp. WAC 7980 and Phoma sp. OMT 5. We report here that these species are probably the same species. The patterns of nucleotide substitutions are more or less dependent upon the percentage of A+T or G+C content. The results contribute to an increasing body of knowledge that recognizes the unsystematic variations among the species, their divergence rate and evolutionary distances at molecular level. Even though we predicted the relative and proportional species contacts, we admit that the molecular experiments should be performed to confirm the accuracy of the predictions.