Anatomy & Physiology: Current Research
Open Access
ISSN: 2161-0940
ISSN: 2161-0940
Research Article - (2023)Volume 13, Issue 2
Background: Chemical Intolerance (CI) is characterized by multi-system symptoms initiated by exposures to environmental toxins. Symptoms include fatigue, headache, mood changes, musculoskeletal pain, gastro-intestinal issues, and difficulties with memory/concentration. With mixed results, researchers have used targeted genetic approaches to understand the genetic pathways associated with CI. This study is the first to apply a genome-wide untargeted exploratory approach.
Methods: A high-density genotyping platform was used to perform a hypothesis-free search for genetic variants associated with CI in a set of 200 participants. Each CI patient was verified using a validated survey. The association between CI and SNPs was obtained using SOLAR (Sequential Oligogenic Linkage Analysis Routines). Gene-chemical-disease interactions were determined using the DisGeNET database.
Results: Several associated SNPs/genes were identified with either increased or decreased risk of CI. Four chemicals were found to alter the gene expressions of the identified SNPs (bisphenol A, valproic acid, aflatoxin B, and benzo (a) pyrene). There were common adverse health effects associated with the genes and the chemicals that influence them. They include inflammation, gastrointestinal and immune system disorders, nervous system diseases, and intellectual disabilities.
Discussion: This study supports evidence of novel genetic components associated with CI that may interact with common ubiquitous chemical and drug exposures affecting gene expression. The identified health consequences are common to individuals with CI and imply gene/chemical exposure interactions that may influence the development or exacerbation of symptoms associated with CI. The identified chemicals affecting these genes are ubiquitous environmental toxins, entering the body through air, food, and water, suggesting the need for greater public health policy efforts.
Chemical; Intolerance; Genes; Chemical exposure; Health effects
Chemical Intolerance (CI) is characterized by multi-systemic symptoms initiated by a one-time high dose or persistent lowdose exposure to environmental toxins. New-onset intolerances often occur when an individual is subsequently exposed to structurally unrelated chemicals, foods, and/or drugs [1]. CI symptoms include fatigue, headache, weakness, rash, mood changes, musculoskeletal pain, gastro-intestinal issues, difficulties with memory, concentration (often described as “brain fog”), and respiratory problems [2-4].
Assessing CI most often involves the use of the Quick Environmental Exposure and Sensitivity Inventory (QEESI), a 50-item validated questionnaire designed to assess intolerances to inhaled chemicals, foods, and/or drugs. The QEESI is used in over a dozen countries around the world and offers high sensitivity and specificity that differentiates CI individuals from the general population [5]. The QEESI is now considered the reference standard for assessing CI and is considered a surrogate for case definition [6,7].
CI prevalence estimates differ by whether it is clinically diagnosed (0.5%-6.5%) or self-reported (average ~20%) in different population based surveys [8-12]. There is evidence of increasing prevalence rates in the US and Japan [5]. According to this research, in just 10 years substantial increases in CI prevalence occurred in both countries. This growth might be attributed to a modern lifestyle and industrialized food consumption.
Increasing numbers of patients and researchers attribute CI to well-defined exposure events, such as indoor air contaminants (e.g., fragranced personal care and cleaning products), exposures to pesticides, new construction or remodeling, or a flood or water damaged building resulting in mold and bacterial growth [13-15]. However, after 50 years, little is known about the physiological and genetic mechanisms that predispose an individual to present with CI [16].
Biological correlates: With mixed results, earlier genetic projects used targeted Single Nucleotide Polymorphism (SNP) association scans focusing on genes involved with phase I/II detoxification, suggesting the need for larger scale genetic projects to investigate the relevance of genetics in CI. As such, various metabolic factors have been investigated. de Luca, et al., present evidence implying that the interplay between alterations in the redox system, glutathione depletion, and pro-inflammatory cytokines affects the expression of metabolizing and antioxidant enzymes in those with CI [17,18]. Subsequently, Dantoft, et al., utilizing immunoassay methods, report supporting evidence for elevated interleukin-1β, IL-2, IL-4, and IL-6, but low IL-13 in CI patients [19]. These results highlight an active role for immune signaling molecules in the pathophysiology of CI. However, subsequent research was not consistent with their earlier findings, and little has been reported [20].
Following this line of exploration, other researchers have used targeted genetic association scans focused on genes that are part of:
• Inflammatory and oxidative stress pathways.
• Genes coding for enzymes that effectively metabolize
xenobiotics (e.g., SOD, NAT),
• Genetic polymorphisms that involve xenobiotic detoxification
processes such as phase I and II enzymes,
• Cytochrome P450s that are responsible for metabolizing
drugs,
• Genes such as PON1/PON2 that are effective in the
metabolization of pesticides.
Targeted genetic scans like these are effective for the confirmation or exclusion of genes that have already been associated with CI [21-27].
Wiesmuller, et al., investigated genetic polymorphisms for 5HTT, NAT1, NAT2, PON1, PON2, and SOD2 genes and found no statistical association between SNPs and CI occurrence [28]. Those results were not consistent with McKeown-Eyssen and coworkers, who did find differences between cases and control. Dantoft, et al., investigated the expression of 26 genes involved in various biochemical pathways including immune regulation, sensory ion channel receptors, serotonin, and receptors for neuromodulators, neural growth factor, and the anti-oxidative enzyme catalase. They found no difference between those with or without CI. Loria-Kohen failed to find specific genetic polymorphisms associated with CI. They did, however, identify specific SNPs (rs1801133 (MTHFR), rs174546 (FADS1) and rs1801282 (PPARγ)) that statistically differed between those with and without CI. Fujimori, et al. targeted specific genotypes and assessed CI using the QEESI in 1,084 employees of Japanese companies. Comparing those with and without CI, no significant differences were found in the allelic distribution of genetic polymorphisms in GSTM1, GSTT1, ALDH2 or PON1 genes [29].
Berg, et al., assert that the inconsistent findings in the literature are primarily caused by gene-environment interactions, genetic heterogeneity in CI, small sample sizes, and/or methodological factors such as the lack of standardized laboratory and/or assessment protocols. Conclusions about the physiological elements associated with CI remain uncertain; although rossi and pitidis point out that there is a greater involvement of the limbic and autonomic nervous system rather than cortical brain processes associated with CI [30]. Further, Micarelli, et al., showed that neurodegenerative factors play a necessary but not sufficient role in CI.
Because of the various inconsistencies of prior studies, Vadala, et al. propose four levels of CI testing to guide clinicians and to further understand CI’s pathology through the combination of multiple methods including quantifiable blood tests, improved diagnostic tools, genetic testing, and thorough clinical observation of symptoms [16].
The identification of new gene variants and the chemicals that effect their expression could potentially increase our understanding about the interaction of genes and specific exposure events that underlie CI. Little has been done toward this end. Our study used a high density genotyping platform to perform a hypothesis-free search for genetic variants associated with CI. Though this work is a hypothesis generating study, we expect that several new or existing genetic variants would be identified that distinguish between those with and without CI. We then sought to identify the interacting chemicals that might influence the expression of those genes.
Sample: Two hundred individuals were recruited as part of the Toxicant-Induced Loss of Tolerance (TILT) program at the University of Texas Health Science Center at San Antonio (UTHSCSA), an environmental health research project designed to improve health outcomes of individuals with CI by identifying environmental triggers in the home and provide best practices for prevention and intervention. Potential respondents were randomly recruited from the waiting room of a family practice clinic and from online solicitation [11]. Participants were at least 18 years old and agreed to the consent agreement approved by the UTHSCSA institutional review board (#HSC20150821H). Each participant completed the QEESI, which is comprised of four 10-item scales measuring chemical intolerance, symptom severity, other intolerances, and life impact, plus the masking index. CI is defined by scores greater than or equal to 40 on both the chemical intolerance and symptom scales. Criteria for control participants were scores less than or equal to 19 on each of these scales. The first 50 clinical respondents who met the criteria for CI and consented to be in the study were retained as cases. Similarly, the first 50 respondents who met criteria for low CI and consented were retained as controls. The same strategy was used for the online recruits to obtain equal numbers of cases and controls from each source.
Sample genotyping: Participants were genotyped using a Infinium global screening array platform (Illumina®) where each participant was genotyped on a set of 654027 independent SNPs. The following quality control procedure was used. High-quality SNPs were selected with MAF (Minor Allele Frequency) ≥0.05, were genotyped in at least 90% of all samples and had a HWE (Hardy-Weinberg equilibrium) ≥10-4. In a complementary way, we filtered samples that were genotyped on at least 90% of all SNPs. Data processing was carried out using PLINK, and the final dataset was composed of 202648 high-quality SNPs genotyped on a set of 188 project participants. The data was stored as a VCF (Variant Calling Format) file on our computer servers [31].
Cryptic genetic relationship estimation: Complete genetic information was obtained for 188 participants and used as input for the calculation of the first 10 genetic Principal Components (PCs). The genetic components were calculated using PLINK, representing a considerable proportion of the unknown genetic relationship between participants. The first three PCs were included in subsequent association analysis to reduce the impact of any unknown genetic relationship among samples.
Genome-wide association scan: The QEESI CI score ranged from 0 to 100, with higher values denoting more severe chemical intolerance. The CI score was inversed normalized and used as a dependent variable of a linear mixed model focused on the genetic variance decomposition as implemented on Sequential Oligogenic Linkage Analysis Routines (SOLAR) [26]. For such a standard pedigree-based variance component model w:
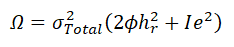
Where;
Ω: Phenotypic covariance matrix,
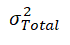
Total phenotypic variance, hr2 , and e2, respectively.
Representing the proportion attributed to the residual additive effect of polygenes, and a random environmental effect. Each genetic variant was tested using a Likelihood Ratio Test (LRT) where the LRT statistic is distributed as a 50:50 mixture of a 1-degree of freedom chi-square and a point of mass zero. All association scans were performed at UTRGV’s computing cluster Medusa.
SNP information: To determine SNP frequency in prior studies, the tissue it is expressed in, and to investigate known clinical significance, we used the national center for biotechnology information database.
Gene-chemical-disease interactions: The set of genes that interacts with chemical response was obtained using DisGeNET database 7.0, a comprehensive gene-disease association database [28,29]. To investigate the interacting chemicals and their effect on genes and disease, we used the comparative toxicogenomic database: Update 2021 [32-34].
Ordinary least squares and logistic regression was used to verify gene/case-status association by regressing the identified gene on chemical intolerance scores or case control status adjusted for age and gender.
Table 1 shows that there were no differences in recruitment method or marital status between cases and controls. Consistent with prior research, we observed a greater percentage of females among the cases compared to controls (88% vs. 12%, p<0.0001), and a significant association between CI and age distribution of those two groups (54 y ± 11.7 vs. 40 y ± 16.2, p<0.001). Significant variables, age, and sex, were used as covariates in our genome-wide association scan genetic models.
| High chemical intolerance N=99 | Low chemical intolerance N=101 | ||
|---|---|---|---|
| Mean (SD) or percentage | Mean (SD) or percentage | ||
| % Female | 88.10% | P<0.001 | 63.60% |
| % Male | 11.90% | 36.40% | |
| Age | 56.3 (11. 7) | P<0.001 | 43.4 (16.2) |
| Marital status | |||
| Married | 43.00% | P<0.30 | 52.10% |
| Divorced/separated | 34.60% | 8.50% | |
| Never married | 14.00% | 35.10% | |
| Widowed/other | 8.40% | 4.30% | |
| Recruitment source | |||
| Clinic | 45% | P<0.51 | 38% |
| Internet | 55% | 62% | |
Table 1: Demographics of study sample.
Participants were genotyped using a commercial Infinium global screening array platform (Illumina®) composed by a set of 654027 independent SNPs. We implemented a strict quality control procedure and defined a set of 20248 high-quality genetic variants genotyped on 188 participants. A set of twelve participants was excluded due to low genotyping efficiency (see methods section for additional details). Each SNP was evaluated for association with the chemical intolerance trait using a linear mixed model as implemented on SOLAR adjusting for age, sex and the first three PCs. Figure 1 shows the distribution of genome-wide association results and compared it with a theoretical null distribution using a QQ-plot (Quantile-Quantile plot) representation.

Figure 1: QQ plot of CHEM trait genome-wide association scan results.
Our top genetic association was the SNP rs62305737 located in an intergenic region between genes LOC101928851 and MIR548AG1 presenting a p value <2.67*10-6 and a high-risk increment of 1.42 standard deviations from the mean. As observed, our association results did not show any evidence of genomic inflation, but no genome-wide significant association was detected (Figure 2).

Figure 2: Manhattan plot of CHEM trait genome-wide association scan results.
This is an expected result due to our small sample size, and we explore our association results even further by focusing on each of the top 50 genetic associations for the CI trait. Initially, we classified SNPs as being of risk or protective for CI, along with the genes flanking each genetic association. For each SNP, we reported their observed empirical minor allele frequency in our study, the frequencies in prior studies, and the percentage of QEESI-assessed CI cases and their mean QEESI chemical score. We also include the tissues where each flanking gene identified is expressed and the clinical significance of the target gene. Table 2 shows that the minor allele frequency of the associated SNPs is highly consistent with prior studies, which serves as a good indication that our samples, despite the small number, are a good representation of the general population. The risk and protective genes, flanking candidate SNPs, are expressed in a wide variety of tissues including neurologic, gastrointestinal, immunologic, and endocrine. To this date, there has not been any reported clinical significance of any of these SNPs.
From Table 2, there is a greater percentage of risk SNPs among those with CI (from 58%-100%) as well as higher QEESI chemical intolerance scores (scores above 40 suggest CI on the QEESI). There is lower percentage of CI among those with the protective SNPs (from 8%-56%) as well as lower QEESI chemical scores (from 7-29).
| SNP frequency | Prior studies frequency | Gene | Tissue expressed in | SNP clinical signifi cance | % CI cases | Mean chem score | % Female | Mean age (SD) | |
|---|---|---|---|---|---|---|---|---|---|
| rs13214731 | 31% | 3-40% | ANKRD6 | Brain ovary | NR | 48/85 56% | 53.6 | 76.0% | 48 (17) |
| rs957788 | 33% | 11-38% | FAM155A | Cerebellum Cortex Frontal lobe | NR | 56/93 60% | 55.6 | 76.4% | 50 (16) |
| rs7194133 | 15% | 11-35% | WWOX | Thyroid Kidney brain | NR | 26/45 58% | 56.6 | 52 (15) | |
| rs985782 | 16% | 6-23% | DAB1 | Small intestine duodena | NR | 34/46 73.9% | 58.7 | 78.3% | 52 (14) |
| rs79060825 | 5% | 1-8% | RIMS2 | Adrenal brain | NR | 10/14 71% | 67.7 | 50.0% | 51 (17) |
| rs75759774 | 5% | 2-6% | DOCK2 | Lymph Appendix Bone Marrow | NR | 11/16 68.7% | 70.8 | 81% | 47 (13) |
| rs56024134 | 3% | 1-3% | SLC25A38 | Thyroid Ovary Bone marrow | NR | 7/8 88% | 78.5 | 89% | 54 (15) |
| rs78933364 | 2% | 1-5% | GIGYF2 | Testis Thyroid ovary | NR | 6/7 86% | 80.65 | 71% | 45 (17) |
| rs78156806 | 2% | 1-6% | BCAS3 | Testis brain | NR | 5/6 83% | 85.9 | 50% | 60 (9.4) |
| rs112808579 | 2% | 1-8% | STK32B | Kidney Testis Stomach | NR | 5/5 100% | 92.4 | 80% | 56 (12) |
| rs183101960 | 2% | 1-6% | ECHDC2 | Liver Fat Ovary | NR | 7/7 100% | 80.54 | 86% | 54 (13) |
| rs78466681 | 2% | 1%-9% | GRIN2A | Cerebellum Cortex Frontal lobe | NR | 1/5 20% | 7.0 | 80% | 55 (12) |
| rs117707051 | 3% | 0.5%-3% | GNA12 | Placenta Ovary | NR | xx | 3.1 | 89% | 48 (14) |
| rs115400407 | 3% | 1%-4% | CXCR2P1 | Appendix Spleen | NR | 2/10 20% | 4.2 | 80% | 47 |
| rs72683492 | 4% | 2%– 5% | LRIG2 | Thyroid Ovary Various | NR | 1/12 8.3% | 6.3 | 72.7% | 45(18) |
| rs2010887 | 30% | 18%– 27% | TNIK | Brain Small intestine Duoden | NR | 34/82 41% | 26.6 | 80.5% | 50 (16) |
| rs1076692 | 21% | 8%-25% | PAK7 | brain | NR | 31/62 50% | 25.2 | 79% | 50 (15) |
| rs9788171 | 37% | 33%-49% | TSPAN9 | Heart Kidney placenta | NR | 42/97 43% | 28.92 | 76% | 48 (15) |
| rs7917473 | 48% | 1%-49% | ARHGAP22 | Brain/thyroid Testis thyroid | NR | 54/118% 56% | 29.04 | 76% | 49 (16) |
Table 2. Summary of SNPS associated with Risk and Protective Effects of Chemical Intolerance
We further explored the set of candidate genes identified on Table 2 by identifying chemicals that directly interact with them by consulting the DisGeNET database. Genes were classified as increasing (risk) or decreasing (protective) the risk of CIs based on the effect coefficient estimate of the genome-wide association scan and were respectively organized in Table 1 (Risk genes) and Table 3 (protective genes). The Tables show the set of interacting chemicals affecting a target gene, and whether those chemicals increase or decrease the expression of the gene. They also show the curated diseases associated with the target gene. Digestive, nervous system, inflammation, affects, neuromuscular and genitourinary issues are apparent across the genes. Figure 3 depicts a heat map representation comparing genes and their interacting chemicals where the interaction of each gene/chemical pair is classified as repressive (-1), positive (+1) or absent (0) (Table 4 and Figure 4).
| Risk genes | Interacting chemicals | Chemical effect on Gene | Diseases associated with gene |
|---|---|---|---|
| ANKRD6 | Valproic acid | ˅/˄ expression | Nervous system diseases learning disabilities/ADHD skin and connective tissue diseases Cardiovascular diseases/CAD Tremor/unsteady gate Inflammation Neurotoxicity syndromes Anxiety disorder |
| Trichostatin A | ˅/˄ expression | ||
| 4-(5-benzo (1,3) dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)benzamide | ˅ expression | ||
| Dorsomorphin | ˅ expression | ||
| Benzo(a)pyrene | ˅ expression | ||
| Bisphenol A | ˅ expression | ||
| Phenylmercuric Acetate | ˅ expression | ||
| 1,4-bis (2-(3,5-dichloropyridyloxy)) benzene | Affects expression | ||
| Abrine | ˅ expression | ||
| Aflatoxin B1 | ˄ Methylation | ||
| FAM155A | 4-(5-benzo (1,3) dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)benzamide | ˅ expression | Digestive system diseases/diverticulitis |
| Dorsomorphin | ˅ expression | ||
| Benzo(a)pyrene | ˄ methylation | ||
| Trichostatin A | ˅ expression | ||
| Valproic acid | ˄ expression | ||
| Aflatoxin B1 | ˅/˄ Methylation | ||
| Copper | ˄ expression | ||
| Dietary Sucrose | ˄ expression | ||
| Entinostat | ˅ expression | ||
| Panobinostat | ˅ expression | ||
| WWOX | Benzo(a)pyrene | ˄ expression/methylation ˅ expression | Ataxia, epilepsy, intellectual disability syndrome due to WWOX deficiency. |
| Aflatoxin B1 | ˄˅ expression | ||
| Valproic acid | ˄ expression | ||
| Bisphenol A | ˄˅ expression | ||
| Sodium arsenite | ˅ expression | ||
| Carbon tetrachloride | ˅ expression | ||
| Decitabine | ˅ expression | ||
| Diethylnitrosamine | ˄˅ expression | ||
| Entinostat | ˄ expression | ||
| Hydrogen Peroxide | ˄˅ expression | ||
| DAB1 | Benzo(a)pyrene | ˄ expression/methylation ˅ expression | Nervous system diseases Mental disorders Skin and connective tissue diseases Hemic and lymphatic diseases; Cardiovascular diseases Tremor Unsteady gate Autism |
| Valproic acid | ˄˅ expression | ||
| 4-(5-benzo(1,3)dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)benzamide | ˅ expression | ||
| Aflatoxin B1 | ˄ ˅ expression/methylation | ||
| Dorsomorphin | ˅ expression | ||
| Pirinixic acid | ˄ ˅ expression | ||
| Trichostatin A | ˅ expression | ||
| Vinclozolin | ˄ ˅ methylation | ||
| Bisphenol A | ˄ ˅ expression | ||
| Carbon tetrachloride | ˅ expression | ||
| RIMS2 | Benzo(a)pyrene | ˄ methylation ˅ expression | Neoplasms respiratory disease Infections Inflammation skin and connective. |
| Valproic acid | ˅ expression | ||
| Trichostatin A | ˅ expression | ||
| 4-(5-benzo(1,3)dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)benzamide | ˄ expression | ||
| Acrylamide | ˅ expression | ||
| Bisphenol A | Affects expression | ||
| Dorsomorphin | ˄ expression | ||
| Magnetite nanoparticles | ˄ expression | ||
| Methamphetamine | ˄ expression | ||
| Pirinixic acid | ˄ expression | ||
| DOCK2 | Benzo(a)pyrene | ˄ methylation ˅ expression | Digestive system diseases Neoplasms Immune system diseases Hemic and lymphatic diseases. |
| Valproic acid | ˄ expression | ||
| Aflatoxin B1 | ˅˄ methylation | ||
| Bisphenol A | ˅ expression | ||
| Cisplatin | ˅ expression | ||
| Entinostat | ˄ expression | ||
| Estradiol | ˄ expression | ||
| (+)-JQ1 compound | ˅ expression | ||
| Rotenone | ˅ expression | ||
| Silicon dioxide | ˄ expression | ||
| SLC25A38 | Tetrachlorodibenzodioxin | ˅ expression | Hemic and lymphatic anemia Nutritional and metabolic diseases Hemic and lymphatic diseases Nervous system Respiratory tract diseases; Nervous system diseases Nutritional and metabolic diseases Iron overload /picks disease |
| 1,2-Dimethylhydrazine | ˄ expression | ||
| Atrazine | ˅˄ expression | ||
| Benzo(a)pyrene | ˅˄ expression | ||
| Acetaminophen | ˅ expression | ||
| Bisphenol A | ˄ expression | ||
| Clofibrate | ˅ expression | ||
| Cyclosporine | ˄ expression | ||
| Doxorubicin | ˅ expression | ||
| Endosulfan | ˄ expression | ||
| GIGYF2 | Ethinyl Estradiol | ˄ expression | Mental disorders Cardiovascular diseases Nervous system disease Sense of smell impaired Frequent falls sleep disorders Anxiety agitation |
| Acetaminophen | ˅˄ expression | ||
| Ozone | ˄ Oxidation | ||
| Tetrachlorodibenzodioxin | ˄ expression | ||
| 7,8-Dihydro-7,8-dihydroxybenzo(a)pyrene 9,10-oxide | ˅˄ expression | ||
| Acrolein | ˄ Oxidation | ||
| ˅ expression | |||
| Aflatoxin B1 | ˅˄ methylation | ||
| Air pollutants | ˅˄ expression | ||
| ˄ Oxidation | |||
| Alpha-pinene | ˄ Oxidation | ||
| ˅ expression | |||
| Bisphenol A | ˄ expression | ||
| BCAS3 | Benzo(a)pyrene | ˅ expression | Female urogenital diseases and pregnancy complications Male urogenital diseases Cardiovascular diseases Gout RBC count Diabetes |
| Bisphenol A | ˅˄ expression | ||
| Aflatoxin B1 | ˄ methylation ˅ expression | ||
| Tetrachlorodibenzodioxin | ˅ expression affects methylation | ||
| Nanotubes, Carbon | ˅˄ expression | ||
| Ozone | ˅ expression | ||
| 1-Methyl-3-isobutylxanthine | ˄ expression | ||
| Acrolein | ˅ expression | ||
| Air pollutants | ˅ expression | ||
| Alpha-pinene | ˅ expression | ||
| STK32B | Benzo(a)pyrene | ˄ expression/methylation ˅ methylation | Nervous system diseases Essential tremor Alzheimer’s Neoplasms CVD Mental disorders GAD. |
| 1-Naphthylisothiocyanate | ˄ expression | ||
| 2,3,7,8-tetrachlorodibenzofuran | ˅ expression | ||
| 4,4'-diaminodiphenylmethane | ˄ expression | ||
| 7,8-Dihydro-7,8-dihydroxybenzo(a)pyrene 9,10-oxide | ˄ expression | ||
| Acrylamide | ˅ expression | ||
| Aflatoxin B1 | ˅ methylation | ||
| Aflatoxin B2 | ˄ methylation | ||
| Atrazine | ˄ expression | ||
| Benzo(e)pyrene | ˄ methylation | ||
| Bisphenol A | ˅ expression | ||
| ECHDC2 | Acetaminophen | ˅ expression | Digestive system diseases Neoplasms Liver carcinoma |
| Aflatoxin B1 | ˄ methylation ˅ expression | ||
| Bisphenol A | ˅˄ expression | ||
| Tetrachlorodibenzodioxin | ˅ expression | ||
| Valproic acid | ˅˄ expression | ||
| Carbon tetrachloride | ˅ expression | ||
| Cisplatin | ˅ expression | ||
| Ethinyl Estradiol | ˅ expression | ||
| 1,2-Dimethylhydrazine | ˅ expression | ||
| Benzo(a)pyrene | ˅ expression |
Table 3: Risk CI Genes and associated interacting chemical and diseases.

Figure 3: Risk genes and their interacting chemicals.
| Protective gene | Interacting chemicals | Chemical effect on gene | Curated diseases associated with gene |
|---|---|---|---|
| GNA12 | Bisphenol A | Increased expression/methylation | Digestive system disease Cognition disorders Prenatal exposure delayed hepatomegaly (enlarged liver) Effects Inflammation Learning disability |
| Valproic acid | Increased expression | ||
| Ozone | Increased oxidation | ||
| Estradiol | Promotor/increased expression | ||
| Ethanol | Increased expression | ||
| Methoxychlor | Increased expression | ||
| Acetaminophen | Increased expression | ||
| Acrolein | Increased oxidation | ||
| Aflatoxin B1 | Increased methylation | ||
| Alpha-pinene | Increased oxidation/abundance | ||
| GRIN2A | Dronabinol (THC) | Decrease expression | Nervous system disease Epilepsy Autism Mental disorder Substance-related disorder Digestive system disease |
| Benzo(a)pyrene | Increase/decrease expression | ||
| Sodium arsenite | Decreased expression | ||
| Bisphenol A | Decreased expression | ||
| Methylmercuric chloride | Decreased expression | ||
| Valproic acid | Increase/decrease expression | ||
| Cocaine | Increased expression | ||
| Lead acetate | Decreased expression | ||
| Vehicle emissions | Increase oxidation/decrease expression | ||
| CXCR2P1 | Triclosan | Increased expression | Congenital abnormalities Fetal growth retardation Skin diseases Prenatal exposure delayed effects Hepatomegaly Inflammation |
| Valproic acid | Increase methylation | ||
| LRIG2 | Benzo(a)pyrene | Increased expression/methylation | Induced liver injury Inflammation Headache Leaning disability Prenatal exposure delayed effects |
| Valproic acid | |||
| Dimethylhydrazine | |||
| Bisphenol A | |||
| Ethinyl estradiol | |||
| Aflatoxin B1 | |||
| Aflatoxin B2 cyclosporine | |||
| TNIK | Valproic acid | Increased expression | Inflammation prenatal exposure delayed effects Induced liver injury Memory and cognition disorders |
| 4-(5-benzo(1,3)dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)benzamide | Increased expression | ||
| Dorsomorphin | Increased expression | ||
| Benzo(a)pyrene | Decrease expression/increased methylation | ||
| Trichostatin A | Increased expression | ||
| Estradiol | Increased expression | ||
| Tetrachlorodibenzodioxin | Decrease/increase expression | ||
| Aflatoxin B1 | Decrease expression/increased methylation | ||
| Belinostat | Increased expression | ||
| Bisphenol A | Increased expression | ||
| PAK7 | Benzo(a)pyrene | Affects methylation | Neurobehavioral manifestations |
| Dietary fats | Affects expression | Learning disabilities | |
| Hexachlorocyclohexane | Increased expression | Prenatal exposure delayed effects Fatty liver Liver neoplasms Kidney diseases Dysbiosis | |
| TSPAN9 | Benzo(a)pyrene | Decrease expression/increased methylation | Weight loss Necrosis Hyperplasia Chemical and drug induced liver injury Hepatomegaly Prenatal exposure delayed effects Liver neoplasms |
| Bisphenol A | Decrease/increase expression | ||
| Clofibrate | Decreased expression | ||
| Acetaminophen | Affects expression | ||
| Ethanol | Increased expression | ||
| Aflatoxin B1 | Decreased methylation | ||
| Benzo(e)pyrene | Increased methylation | ||
| Estradiol | Decreased expression | ||
| Methapyrilene | Increased methylation | ||
| Pirinixic acid | Decreased expression | ||
| ARHGAP22 | Bisphenol A | Decrease/increase expression | Necrosis Weight Loss Chemical and drug induced liver injury Inflammation Prenatal exposure delayed effects Hepatomegaly Kidney diseases Hyperplasia Fibrosis |
| Benzo(a)pyrene | Increase/decrease expression, methylation | ||
| Valproic acid | Increased expression and methylation | ||
| Cisplatin | Decrease/increase expression | ||
| Dexamethasone | Decreased expression | ||
| Aflatoxin B1 | Increased expression | ||
| Dietary fats | Decreased and increased expression | ||
| Ethinyl estradiol | Increased expression | ||
| Jinfukang | Increased expression | ||
| Methylcholanthrene | Increased expression |
Table 4: Protective CI genes and associated interacting chemical and diseases.

Figure 4: Protective genes and their interacting chemicals.
The most common chemicals interacting with most genes among both the risk and protective genes are valproic acid, benzo(a)pyrene, aflatoxin B1, and bisphenol A.
Other chemicals affecting at least 3 risk genes are tetrachlorodibenzodioxin, acetaminophen, ethinyl estradiol, dorsomorphin, trichostatin A, carbon tetrachloride, and entinostat. Only estradiol affects 3 genes among the Protective genes. The remaining chemicals for both set of genes affect 2 or fewer genes.
In Table 5, we included all the risk SNPs (model 1) in an ordinary least squares regression model as predictors of the QEESI chemical intolerance score. Genes are coded from 0 to 2 and vary by individual, representing the number of minor alleles observed for that specific SNP genotyped on each participant (ranging from 0-8). Of the risk SNPs/genes, four were significantly and positively associated with CI scores: rs957788/FAM155A, rs985782/DAB1, rs56024134/SLC25A38, and rs112808579/STK32B. Each contributed to a 12-to-42-point increase in the QEESI chemical intolerance score per one unit increase of the minor allele count.
In model 2, the protective SNP/genes were used to predict QEESI chemical intolerance scores. Four genes were significantly and inversely associated with QEESI chemical intolerance scores: rs117707051/GNA12, rs2010887/TNIK, rs9788171/TSPAN9, and rs7917473/ARHGAP22. These genes were associated with an 8 to 26 points decrease in the chemical intolerance score (Table 5).
| Model 1: Risk SNPs/genes | Model 2: Protective SNPs/genes | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP/gene | Parameter estimate (standard error) | p | STD | SNP/gene | Parameter estimate (standard error) | p | STD | ||
| rs13214731 ANKRD6 | 3.84 | -3.87 | 0.321 | 0.07 | rs78466681 GRIN2A | -5.48 | 15.85 | 0.72 | -0.03 |
| rs957788 FAM155A | 12.49 | -3.88 | 0.001 | 0.22 | rs117707051 GNA12 | -26.35 | 11.98 | 0.02 | -0.17 |
| rs7194133 WWOX | 3.44 | -4.65 | 0.46 | 0.05 | rs115400407 CXCR2P1 | -7.67 | 10.9 | 0.482 | -0.05 |
| rs985782 DAB1 | 23.02 | -4.99 | <.0001 | 0.33 | rs72683492 LRIG2 | -14.57 | 9.81 | 0.13 | -0.11 |
| rs79060825 RIMS2 | 10.46 | -8.43 | 0.21 | 0.09 | rs2010887 TNIK | -11.53 | 3.79 | 0.002 | -0.22 |
| rs75759774 DOCK2 | 5.42 | -8.22 | 0.51 | 0.05 | rs9788171 TSPAN9 | -8.9 | 3.77 | 0.01 | -0.17 |
| rs56024134 SLC25A38 | 37.17 | -11.85 | 0.002 | 0.23 | rs7917473 ARHGAP22 | -8.09 | 3.75 | 0.03 | -0.16 |
| rs78933364 GIGYF2 | 4.07 | -12.89 | 0.75 | 0.02 | |||||
| rs78156806 BCAS3 | -7.97 | -14.6 | 0.58 | -0.04 | |||||
| rs112808579 STK32B | 41.94 | -14.9 | 0.0055 | 0.2 | |||||
Table 5: OLS regression: Individual risk genes predicting QEESI chemical intolerance.
In Table 6, both sets of genes were entered into the model simultaneously. There were 4 remaining significant risk genes: FAM155A, SLC25A38, DAB, and STK32B. Each contributed to a 9-to-34-point increase in the QEESI chemical intolerance score per one unit increase of the minor allele count. Only one protective gene, ARHGAP22, was significantly associated with an 8-point decrease in CI scores, and TNIK was of marginal significance (p<0.06), with a 7-point decrease in CI scores.
| Model 3: SNP/gene | ||||
|---|---|---|---|---|
| Risk SNPs | Parameter estimate | Standard error | p | Standardized estimate |
| Intercept | 38.7 | -6.58 | <.0001 | 0 |
| rs13214731 | 2.57 | -3.79 | 0.49 | 0.04 |
| rs957788 FAM155A | 9.51 | -3.98 | 0.01 | 0.16 |
| rs7194133 | 1.26 | -4.54 | 0.78 | 0.02 |
| rs985782 DAB1 | 21.33 | -4.97 | <.0001 | 0.3 |
| rs79060825 | 5.15 | -8.36 | 0.53 | 0.046 |
| rs75759774 | 5.74 | -7.98 | 0.47 | 0.048 |
| rs56024134 SLC25A38 | 33 | -11.53 | 0.004 | 0.2 |
| rs78933364 | -2.41 | -12.58 | 0.84 | -0.01 |
| rs78156806 | -2.52 | -14.36 | 0.86 | -0.01 |
| rs112808579 STK32B | 33.76 | -14.56 | 0.02 | 0.16 |
| rs78466681 | -3.52 | -14.032 | 0.8022 | -0.01 |
| rs117707051 | -17.2 | -10.75 | 0.112 | -0.11 |
| rs115400407 | -6.76 | -9.81 | 0.4919 | -0.04 |
| rs72683492 | -4.25 | -8.84 | 0.6313 | -0.03 |
| rs2010887 TNIK | -6.67 | -3.54 | 0.0618 | -0.12 |
| rs9788171 | -4.51 | -3.56 | 0.2072 | -0.08 |
| rs7917473 ARHGAP22 | -7.85 | -3.43 | 0.0237 | -0.15 |
Table 6: OLS regression: Individual risk and protective genes together predicting QEESI chemical intolerance scores.
The top of Figure 5 shows that those with CI have a higher total number of risk genes on average than those without CI (3.2 vs. 1.9 respectively). Further, those with CI have fewer protective genes than those without CI (1.6 vs. 2.9 respectively). Figure shows the separate probability plots of the risk and protective genes from a logistic model where SNPs are predictors of QEESI-defined CI. Among the risk SNPs, for each increase in the number of alleles, there is a corresponding 1.8 odds ratio (95% CI=1.4-2.5) representing an exponential increase in the probability of CI. With 4 alleles the probability of CI reaches 75% and increases further with each additional gene. The odds ratio for the protective SNPs was 0.5 (95% CI=0.3-0.6). This represents a 50% reduced probability of CI with each additional protective SNP. With each additional protective SNP, there is rapidly diminishing probability of CI.

Figure 5: Logistic regression: number of risk and protective genes predicting CI status, adjusted for gender and age.
We identified several novel SNPS/genes through a hypothesis-free, untargeted GWAS approach, confirming our initial supposition. The SNPs led to the identification of genes that were classified as risk or protective based on the direction of effect of the regression analysis. Prior studies have used a targeted gene analysis, and to our knowledge, this is the first time a GWAS was performed focused on CI. CI prevalence follows a standard polygenic model of inheritance where variable, but additive environmental and genetic components exist. However, the SNPs identified by our study have not been previously reported. There is a positive correlation between CI status and the total number of risk genes an individual carries. Conversely, individuals carrying greater numbers of protective genes have a lower occurrence of CI risk. These findings suggest a linear association between the number and type of genes an individual possesses and their CI diagnostic. When both the risk and protective genes were simultaneously entered into a logistic regression model, most of the risk genes persist as significant predictors of CI, and only one protective gene remained significant. This result indicates that possessing those risk genes are more of a hindrance despite possessing protective genes. The comprehensive sets of protective and risk genes were used as input to consult the DisGeNET database. We identified several candidate chemicals that could directly interfere with the gene expression control of those genes. From Figures 3 and 4, the chemicals affecting the greatest number of both risk and protective genes were valproic acid, benzo (a) pyrene, aflatoxin B1, and bisphenol A. Several health effects and diseases are associated with these chemicals that are common to the effects of the genes they influence.
Valproic Acid (VPA) is a widely used anti-convulsant, mood stabilizer, and migraine headache medication, common symptoms of VPA toxicity include altered cognitive function, depression, hepatic injury, skin rash, and gastrointestinal issues [35,36]. Clinical studies have demonstrated that in utero exposure to VPA is associated with birth defects, cognitive deficits, and increased risk of autism [37]. This is relevant because prior research shows that mothers with CI are at three times the risk for having children with autism or ADHD [38]. Further the health effects associated with both the protective and risk genes in Tables 3 and 4 correspond with the side effects of VPA. For example, the six protective genes that VPA influences from Figure 3 are LRIG2, ARHGAP22, TNIK, CXCR2P1, GRIN2A, and GNA12. The common health issues associated with these genes, as presented in Table 4, demonstrate a common theme, and are related to the side effects of VPA: Child development effects, inflammation, cognitive influences, and liver function issues often experienced by those with CI. VPA also influences 7 risk genes in Figure 4: DAB1, ANKRD6, RIMS2, FAM155A, DOCK2, ECHDC2, and WWOX. Again, common health issues associated with these risk genes and VPA toxicity share key health issues, including digestive system diseases, neoplasms, nervous system diseases, intellectual disabilities including autism, and immune system diseases/inflammation.
In Table 5, we included all the risk SNPs (model 1) in an ordinary least squares regression model as predictors of the QEESI chemical intolerance score. Genes are coded from 0 to 2 and vary by individual, representing the number of minor alleles observed for that specific SNP genotyped on each participant (ranging from 0-8).
In previous studies we have demonstrated the connection between mast cell proliferation and chemical intolerance, as well as chemical intolerance and autism Interestingly, Shin, et al. demonstrated that BALB/c hairless mice (a standard mouse model for autism) exposed to VPA, showed increased activity of cytokine markers as well as mast cell proliferation in skin and brain. They propose that this shared susceptibility of the brain and epidermis to the known neurotoxicity of VPA, suggests that the atopic diathesis (e.g., immune mediated dermatitis) could be extended to include autism [39,40].
Bisphenol A (BPA) is a ubiquitous environmental chemical produced in copious quantities for use in the production of plastics for products in everyday use such as water bottles and water supply pipes and is in a large portion of the world’s food supply. It is also an unavoidable contaminant of corn-based animal feed [41]. BPA enters the body through the digestive and respiratory tracts as well as via skin absorption. Food and beverages account for most daily human exposure [42]. BPA is a liver carcinogen and endocrine disruptor causing damages to the reproductive, immune, and neuroendocrine systems with consequences such as growth impairment, malnutrition, and immunomodulation [43]. The protective genes that BPA influence are the same as mentioned above and have shared gene/chemical health effects including child developmental effects, cognitive influences, inflammation, and liver function/disease. Again, these are common comorbidities associated with CI [44].
BPA affects all the risk genes except FAM155A and includes the aforementioned shared health effects.
Aflatoxin B1 (AFB1): Aflatoxins are carcinogenic mycotoxins produced by Aspergillus fungi and are known to contaminate a substantial portion of the world's food supply. Aflatoxin B1 is the most potent of the aflatoxin compounds and a well-known liver carcinogen in humans and animals. It is also associated with growth suppression, malnutrition, immunomodulation, mental impairments, and GI pain [45]. From Figures 3 and 4, AFB1 affects the same protective genes as VAP and BPA. Similarly, AFB1 influences most of the same risk genes. The shared health effects mentioned above are therefore consistent.
Benzo (A) pyrene (BP) is also a ubiquitous environmental contaminant found in foods and air. It is a coal tar polyaromatic hydrocarbon and a known carcinogen as well as affecting lung function (e.g., bronchitis) [46,47]. BP exposure has neurobehavioral effects and is a developmental neurotoxin affecting reproductive and immunological function. Adverse birth outcomes include reduced birth weight, postnatal body weight, and head circumference [48,49]. Sources of exposure include vehicle exhaust emissions, heat and power generation, refuse burning, industrial processes, oil contamination by disposal or spills, cigarette smoke, and cooking of foods [50,51]. Again, as shown in Figures 3 and 4, BP influences most of the same risk and protective genes as AFB1, BPA and VPA, and shares many of the same health effects.
This study supports the existence of novel genetic components associated with CI that may interact with common chemical and drug exposures affecting gene expression, with potential chronic disease and other adverse health consequences. All candidate genes identified in this project need to be further independently validated to ensure the replication of our initial findings [52-58].
We identified novel SNPs that differentiate those with and without CI. The known health effects linked with the genes were similar to the health effects associated with the chemicals which influence the genes. Those shared health effects are common among individuals diagnosed with CI. This implies that important gene/chemical exposure interactions may influence the development or exacerbation of the health effects associated with CI. Up or down-regulated disruption of the genes by these chemicals may influence the severity of symptoms among individuals with CI. We also found that the more risk genes an individual had, the greater the likelihood of CI. Conversely, the more protective genes, CI was less likely. When both sets of genes were simultaneously entered in a logistic model, many of the risk genes remained significant predictors of CI, while all but one protective gene remained. This implies that when both types of genes compete, the ones associated with risk take precedence. Three of the four most influential chemicals that affect the genes in this study are ubiquitous environmental toxins, entering the body through air, food, and water; suggesting the need for greater coordination between air, food, and water quality mitigation policies particularly among the growing number of vulnerable individuals with CI. The results of this study could play a significant role in a new generation of genetic projects focused on the study of CI with more diverse human populations exposed to different environmental conditions.
This study had a relatively low sample size for a genetic study. As such, statistical power was reduced, and Bonferroni correction was not used. Rather, we interpretated the data favoring biological relevance of the candidate genes that we found predicted the CI status of participants. We found SNPs that have yet to be identified as clinically relevant. Therefore, an independent validation in other population communities with larger samples and better case-control matching is also warranted.
This research was made possible by the generous support of the Marilyn Brachman Hoffman Foundation.
All authors contributed to the study conception and design. Material preparation, Roger B. Perales and Rudy Rincon performed data collection. Genetic analyses were performed by Marcio Almeida and overall statistical analysis was performed by Ray Palmer. Ray Palmer and Marcio Almeida wrote the first draft of the manuscript. All authors contributed to the revisions of the manuscript and approved the submitted version.
This study was approved by the university of Texas health science center San Antonio internal review board (approval number HSC20150821H).
The datasets of the current study are available from the corresponding author on reasonable request.
The authors declare that they have no competing interests.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Palmer RF, Almeida M, Perales RB, Rincon R, Miller CS (2023) An Untargeted Genome-Wide SNP Investigation of Chemical Intolerance. Anat Physiol. 13:426
Received: 11-Feb-2023, Manuscript No. APCR-23-21777; Editor assigned: 13-Feb-2023, Pre QC No. APCR-23-21777 (PQ); Reviewed: 27-Feb-2023, QC No. APCR-23-21777; Revised: 11-Apr-2023, Manuscript No. APCR-23-21777 (R); Published: 18-Apr-2023 , DOI: 10.35248/2161-0940.23.13.426
Copyright: © 2023 Palmer RF, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.