Virology & Mycology
Open Access
ISSN: 2161-0517
ISSN: 2161-0517
Research Article - (2022)Volume 11, Issue 1
The high impact of diseases caused by dengue viruses on global health is now reflected in an increased interest in the identification of drug targets and the rationale-based development of antiviral inhibitors which are suitable for a causative treatment of severe forms of dengue virus infections dengue haemorrhagic fever and dengue shock syndrome. A promising target for the design of specific inhibitors is the dengue virus NS3 serine protease which in the complex with the small activator protein NS2B-catalyses processing of the viral polyprotein at a number of sites in the nonstructural region. The NS3 protease is an indispensable component of the viral replication machinery and inhibition of this protein offers the prospect of eventually preventing dengue viruses from replication and maturation.
To this effect, several computational approaches were applied in this work. Initially molecular docking studies of reference ligands to the DEN3 NS2B/NS3 serine protease were carried out. These reference ligands consist of reported competitive inhibitors, 4-hydroxypanduratin A and panduratin A, screened from natural products database and 17 designed ligands GSP1, GSP2, GSP3, GSP4, GSP5, GSP6, GSP7, GSP8, GSP9, GSP10, GSP11, GSP12, GSP13, GSP14, GSP15, GSP16 and GSP17. In the designing of new lead inhibitors, the enzyme complexed to the reference ligands was minimized and their complexation energies (i.e., sum of interaction energy and binding energy) were computed. New compounds as potential dengue inhibitors were then designed by putting various substituents successively on the benzyl ring A of the reference molecule. These substituted benzyl compounds were then computed for their enzyme-ligand complexation energies. New enzyme-ligand complexes, exhibiting the lowest complexation energies and closest to the computed energy for the reference compounds, were then chosen. Thus, GSP6 is proposed as a potential inhibitor to the NS3/NS2B protease activities of DEN-3 with the lowest complexation energy of -92.4 kcal/mol. The design of drugs to treat dengue is at a very exciting crossroads, not only because scientific progress on DENV targets has been tremendous in the last five years, but also because the dengue problem is huge and is increasing, such that dengue might soon join the list of diseases with economically viable drug markets.
DEN-3 NS2B/NS3 protease; Molecular docking; Interaction energy; Binding energy; Complexation energy
Dengue virus is a major threat to health in tropical countries around the world. It is limited primarily to the tropics because a tropical mosquito transmits it, but even with this limitation, 50-100 million people are infected each year. Most infected people experience dengue fever, with terrible headaches and fever and rashes that last a week or two. In some cases, however, the virus weakens the circulatory system and can lead to deadly hemorrhaging. Researchers are now actively studying the virus to try to develop drugs to cure infection, and vaccines to block infection before it starts.
Genome organization
Dengue viruses, members of the Flaviviridae family, possess singlestranded, positive sense RNA genomes and generate mature viral proteins by co- and post-translational proteolytic processing of a polyprotein precursor catalyzed by host cell and virus encoded proteases. The genomic RNA of dengue virus serotype 3 contains 10,723 nucleotides and encodes a single polyprotein precursor of 3,391 amino acid residues [1]. Individual viral proteins are arranged in the order C-prM-E-NS1-NS2A-NS2B-NS3-NS4A- NS4B-NS5. Proteolytic cleavages in the N-terminal region of the viral polyprotein are mediated by a host signal peptidase and yields three structural proteins C, prM and E, which constitute the virion [2]. Before the virion exits the cell, prM is cleaved by a cellular furin-type prohormone convertase in the post-Golgi acidic compartment to yield the M protein [3]. Cleavages at the NS1/ NS2A and NS4A/NS4B junctions are catalyzed by a signalase bound to the membranes of the ER [4,5]. Proteolytic cleavages in the nonstructural region of the polyprotein are mediated by a heterodimeric complex of NS3 with the activator protein NS2B which catalyses in cis (intramolecular) cleavages at the NS2A/ NS2B and NS2B/NS3 sites and in trans (intermolecular) cleavages at the NS3/NS4A and NS4B/NS5 polyprotein junctions [6-8]. Additional cleavages mediated by the NS3 protease within the C, NS2A, NS4A and within a conserved C terminal portion of NS3 itself have been described in the literature [7-9]. His Cleavage sites recognized by the NS2B/NS3 protease consist of ‘dibasic’ residues Lys-Arg, Arg-Arg and Arg-Lys at the (nonprime) P1 and P2 positions of the cleavage site sequence followed by short chain residues such as Gly, Ala and Ser at the (prime) P1’ position as shown in Figure 1.

Figure 1: Schematic representation of dengue virus polyprotein processing (Sites on the flavivirus polyprotein cleaved by host- encoded proteases and the virus-encoded two-component protease NS2B-NS3).
The NS3 protease domain
The existence of a trypsin-like serine protease domain in the N-terminal region of the flaviviral NS3 proteins was originally predicted by sequence comparisons between cellular and virus- encoded proteases [10]. The NS2B-NS3 endopeptidases of the Flavivirus genus which at present comprises at least 68 known members, are now commonly designated as flavivirin (EC 3.4.21.91) [11,12]. The dengue virus 69 KDa NS3 protein is a multifunctional protein with a serine protease domain located within the N-terminal 167 amino acid residues [13] and activities of a nucleoside triphosphatase (NTPase) and RNA helicase in the C-terminal moiety [14]. A catalytic triad consisting of residues His, Asp and Ser was identified by site-directed mutagenesis experiments and replacement of the catalytic serine by alanine resulted in an enzymatically inactive NS3 protein [15]. The NS3 protease is an essential component for maturation of the virus and viable virus was never recovered from infectious cDNA clones carrying mutations in the NS3 sequence which abolished protease activity [16]. Interaction of the helicase portion of NS3 with the viral RNA dependent RNA polymerase NS5 may promote the association of the viral replicase complex to the membranes of the ER [17]. The 3-dimensional structure of the N terminal 185 residues of the dengue virus NS3 protease domain (NS3pro) was resolved at a resolution of 2.1 Å [18]. The overall folding of the protein resembles the 6-stranded b-barrel conformation typical for chymotrypsin-like serine proteases. Interestingly, the structure of the dengue virus NS3 protease is closer to that of the hepatitis C virus NS3/NS4A co-complex than to the unliganded HCV NS3 protease, an observation which is suggestive of major structural differences in the co-factor dependent activation mechanism of the two proteases [19]. The substrate binding site of NS3pro is relatively shallow and contourless and specific enzyme-substrate interactions were not predicted to extend beyond the P2 and P2’ positions of the substrate peptide in the absence of the NS2B cofactor [20].
The NS2B co-factor
The presence of a small activating protein or co-factor is a prerequisite for optimal activity of the flaviviral NS3 proteases with their natural polyprotein substrates. Although the dengue virus NS3 protease exhibits NS2B independent activity with model substrates for serine proteases, enzymatic cleavage of dibasic peptides is markedly enhanced with the NS2B-NS3 co-complex and the presence of the NS2B activation sequence is indispensable for the cleavage of polyprotein substrates in vitro [21]. The initial characterization of the co-factor requirement for the dengue virus NS3 protease had revealed that the minimal region necessary for protease activation was located in a 40-residue hydrophilic segment of NS2B [22]. The hydrophobic flanking regions of the 14 KDa NS2B proteins are likely to be involved in targeting the protease complex to the membranes of the ER where genome replication occurs. Fusion of the NS2B core sequence to the NS3 protease domain yielded a catalytically active NS2B(H)-NS3p protein, which, upon expression in E. coli and subsequent refolding, displayed autoproteolytic processing at the NS2B/NS3 site conducive to the formation of a non-covalent adduct [23]. Incorporation of a flexible nonamer linker, Gly4-Ser-Gly4, between the NS2B core segment and the protease domain resulted in a cleavage-resistant protease with optimized enzymatic activity against hexapeptide substrates representing native polyprotein cleavage junctions [24]. A recombinant construct representing the full length NS2B co- factor linked to the NS3 protease domain was enzymatically active with peptide substrates derived from the polyprotein; however, this protein was completely resistant to proteolysis self-cleavage [24].
Objective
• Molecular docking studies of NS2B/NS3 protease with its inhibitors and to find out the most potent inhibitor against DEN3 NS2B/NS3 PROTEASE COMPLEX via molecular docking studies.
• To unravel the various interactions between inhibitors and residues in NS2B/NS3.
• To design new ligands having lowest complexation energy that will act as inhibitors against NS2B/NS3 PROTEASE complex.
Ligand structures optimization
The ligand structures were minimized with Hyperchem Pro 8.0 software using conjugate gradient Polak-Ribiere method to obtain the most stable structure geometry.
Preparing enzyme structure
The NS2B cofactor is essential for the protease catalytic activity of NS3. The sequences of NS2B (gi|25992094|gb|AAN77048.1|) and NS3 (gi|156619339|gb|ABU88349.1|) protease were retrieved from NCBI. Their three dimensional structure was determined individually via GENO-3D.
Enzyme structure refinement and optimization
The enzyme structures were minimized according to energy criteria with the KOBAMIN software. This procedure was set up to bring the energy level of the system to minimum and structurally stable.
Protein-protein docking
The two protein structures were docked to form a protein-protein complex by using GRAMMX software and the complex structure was again refined and optimized.
Catalytic triad identification
The catalytic triad (Asp-Ser-His) was identified at the active site 4 of the protease complex using Pocket finder software.
Protein ligand molecular docking
The docking of ligands to the catalytic triad of NS3-NS2B protease was performed using Docking Server. Docking server is reported to be the most popular docking program and is reliable. Using the software, polar hydrogen atoms were added to the enzyme and its nonpolar hydrogen atoms were merged, whereas for the ligand, nonpolar hydrogen atoms were merged and Gasteiger charges were added. All rotatable bonds of ligands were set to be rotatable. All calculation for protein-fixed ligand-flexible docking was done using the Lamarckian Genetic Algorithm (LGA) method. A population size of 150 and 10 million energy evaluations were used for 100 search runs. The grid box with a dimension of 60 x 60 x 60 points and 0.375 Å grid spacing was used around the catalytic triad to cover the entire enzyme binding site and accommodate ligands to move freely. After the docking searches were completed, clustering histogram analysis was performed based on an RMSD (root mean square deviation) of not more than 1.5 Å. The best conformation was chosen from the most populated cluster with the lowest docked energy. The interactions of complex enzyme-ligand conformations, including hydrogen bond and other interactions, were analysed using PYMOL.
The sequences of DEN-3 NS2B (gi|25992094|gb|AAN77048.1|) and NS3 (gi|156619339|gb|ABU88349.1|) protease were obtained from NCBI.
3D model of DEN-3 NS2B/NS3
The 3D model was obtained by using GENO-3D software and the region surrounding the catalytic triad of the protease and the residues involved in the substrate binding showed a good level of sequence identity. The template for NS2B used was: 3E90 (PDB-ID) and template for NS3 was 1YKS (PDB-ID). In addition, the results obtained from PROVE, VERIFY3D and ERRAT revealed the good quality of the model built as shown in Figures 2-4.

Figure 2: 3D structure of NS2B protein.

Figure 3: 3D structure of NS3 protein.

Figure 4: 3D structure of NS2B/NS3 protease complex using GRAMMX.
The catalytic triad
The active sites present in the protein complex were predicted by using POCKETFINDER software. The catalytic triad of the serine protease was present in the active site 4 as shown in Figures 5 and 6 below:

Figure 5: Active sites predicted (The red colored active site contains the catalytic triad of serine protease).

Figure 6: Site 4 having catalytic triad (closer view).
Predicted site 4
Site Volume: 138 Å3
Protein Volume: 11503 Å3
Inhibition of bioactive compounds towards DEN-3 NS2B/ NS3
Docking of 4-Hydroxypanduratin A and Panduratin A to DEN-3 NS2B/NS3 several competitive inhibitors towards Dengue serine protease activity have been discovered through a substrate-based approach by mimicking the polyprotein cleavage junctions. In addition, α-keto peptidomimetic compounds and guanidine derivatives have also been targeted as competitive inhibitors towards the Dengue serine protease. The natural products, 4-hydroxpanduratin A and panduratin A were found to competitively inhibit the activity of the DEN-3 serine protease. The structures of these compounds are shown in Figures 7a and 7b. The complexation energy for both competitive inhibitors were calculated and summarized in Table 1. 4-hydroxypanduratin A has a lower complexation energy value compared to panduratin A.

Figure 7: (a) Structure of 4-hydroxypanduratin A, (b) Structure of panduratin A.
| Molecule | Ki (µM) | Complexation energy (kcal/mol) |
|---|---|---|
| 4-hydroxypanduratin A | 25 | -69.2 |
| Panduratin A | 29 | -56.7 |
Table 1: The complexation energies of 4-hydroxypanduratin A and panduratin A.
Two competitive dengue inhibitor compounds were docked onto the serine protease enzyme. Figure 8 shows the superimposition of these two compounds (i.e. 4-hydroxypanduratin A and panduratin A). Both 4-hydroxypanduratin A and panduratin A were observed to take up similar poses with similar binding orientation around the active sites of the serine protease NS2B/NS3 (Figures 9a and 9b) and these ligands were observed to interact with the residues in the catalytic triad (i.e., His70, Asp77 and Ser68) of the protease.

Figure 8: Superimposition of the competitive inhibitor structures, 4-hydroxypanduratin A (yellow) and panduratin A (orange).

Figure 9: (a) Docked structures of 4-hydroxypanduratin A, (b) Docked structures of panduratin A.
Design of new dengue virus inhibitors based on 4-hydroxypanduratin A and panduratin A
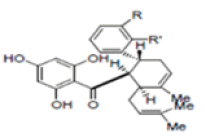
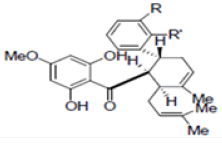
New compounds with potential inhibitory activities against dengue virus were then designed by adding substituent’s on the various positions (i.e. positions 1, 2, 3, 4 and 5) of the benzyl ring A for 4-hydroxypanduratin A and panduratin A (Figure 9a) since this ring gave the highest contribution and has many possibilities for substitution. In this work, polar substituents such as –OH, carbonyl group (i.e. –COO-), nitro group (i.e. -NO2 ), amine group (-NH3 +), alkyl group (i.e. -CH3 ) and halogen group (i.e. –Cl) were placed on positions 1 to 5 of the benzyl ring A. These substituents were added one by one on each position of the ring as shown in Figure 10.

Figure 10: Molecular structures of competitive inhibitors extracted from Boesenbergia rotunda. Note: Panduratin A (R: OMe; R’: OH); 4-hydroxypanduratin A (R: OH; R’: OH)
The complexation energies for these newly designed derivatives are presented in Table 2. The derivatives for the ligands substituted at positions 4 and 5 were observed to have complexation energy relatively close to that of 4-hydroxypanduratin A and panduratin A (i.e. -69.2 kcal/mol for 4-hydroxypanduratin A and -56.7 kcal/mol for panduratin A) are presented in Table 3. The designed ligands (GSP5 and GSP8) with lowest complexation energy as shown in Figures 11.

Figure 11: GSP8 (-70.1 kcal/mol).
| 4-hydroxypanduratin A | ||||||
|---|---|---|---|---|---|---|
| Position in benzyl ring A | Complexation energy of derivatives (kcal/mol) | |||||
| -OH | -COO- | -NO2 | -NH3+ | -Cl | -CH3 | |
| 1 | -35.1 | 60.1 | -1.6 | -49.1 | -43.1 | -38.6 |
| 2 | -50.9 | 32.2 | 242.6 | -36.5 | -50.7 | -44.0 |
| 3 | -27.8 | 54.2 | -49.3 | -63.3 | -53.8 | -43.9 |
| 4 | -41.7 | -22.8 | -68.7 | -68.3 | -41.4 | -38.5 |
| 5 | -35.1 | -90.4 | -44.4 | -93.9 | -61.6 | -64.1 |
| Panduratin A | ||||||
| Position in benzyl ring A | Complexation energy of derivatives (kcal/mol) | |||||
| -OH | -COO- | -NO2 | -NH3+ | -Cl | -CH3 | |
| 1 | 72.9 | 13.1 | 59.9 | 121.9 | 52.8 | 64.4 |
| 2 | 68.5 | 36.9 | -24.8 | -14.4 | -31.6 | -37.9 |
| 3 | -23.7 | 5.9 | -48.4 | -11.7 | -62.2 | -62.6 |
| 4 | -51.7 | 27.8 | -41.6 | -42.7 | -59.1 | -53.7 |
| 5 | -60.8 | -44.7 | -58.3 | -54.6 | -55.2 | -54.0 |
Table 2: Complexation energies of some potential inhibitor derivatives.
| Sl No. | Molecule | R | R' | Complexation energy (kcal/mol) |
|---|---|---|---|---|
| 1 |
|
R=NO2 | R'=H | -68.5 |
| R=NH3+ | R'=H | -69.1 | ||
| R=CH2NH3+ | R'=H | -69.6 | ||
| R=NH2+CH3 | R'=H | -69 | ||
| R=H | R'=COO- | -91.6 | ||
| R=H | R'=NH3+ | -92.4 | ||
| 2 |
|
R=Cl | R'=H | -68.1 |
| R=OCH3 | R'=H | -70.1 | ||
| R=SH | R'=H | -57.8 | ||
| R=NH2+CH3 | R'=H | -65.8 | ||
| R=H | R'=OH | -61.8 | ||
| R=H | R'=NO2 | -58 | ||
| R=H | R'=NH3+ | -64.7 | ||
| R=H | R'=Cl | -56.2 | ||
| R=H | R'=CH2OH | -66.1 | ||
| R=H | R'=SH | -63.3 | ||
| R=H | R'=CH2NH3+ | -58.6 |
Table 3: Some 4-hydroxypanduratin A and panduratin A derivatives substitution at the 4 and 5 positions of the benzyl ring A (R and R’ respectively).
The docking of various ligands with competitive activities to the model of DEN3 NS3 serine protease complexes with NS2B co-factor was carried out. Several modes of interactions such as H-bonding, Vander Waals as well as π-π interaction were observed between these ligands and the DEN-3 NS2B/NS3 active sites. These findings have provide further understanding on the binding interaction of the catalytic triad of the DEN-3 NS2B/NS3 serine protease, thus giving input into the mode of action of the catalytic triad. In this study, we designed 17 ligands (inhibitors) against DEN-3 NS2B/NS3 protease namely GSP1, GSP2, GSP3, GSP4, GSP5, GSP6, GSP7, GSP8, GSP9, GSP10, GSP11, GSP12, GSP13, GSP14, GSP15, GSP16, GSP17. The GSP6 was observed as the best protease inhibitor candidate, with value of complexation energy -92.4 kcal/mol. Thus, the GSP6 is a potential inhibitor drug candidate to the NS2B/NS3 protease activities of DEN-3 apart from already existing inhibitors like 4-hydroxypanduratin A and panduratin A (i.e. -69.2 kcal/mol for 4-hydroxypanduratin A and -56.7 kcal/mol for panduratin A). If this strategy of GSP6 used for further drug development, it can be a remarkable achievement.
The theoretical models will further pave way to experimentally lead in pharmacogenomics and drug development. The data (GSP6) which are available, now, make the dengue virus NS3 protease a valid molecular target for the development of antiviral compounds. If such kind of computer aided drug designing approach is consider there would be a remarkable achievement in the upcoming future to overcome the threat of Dengue virus-3.
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
[Crossref], [Google Scholar]
Citation: Gware P, Gupta G (2022) Analogues of 4-Hydroxypanduratin-A and Panduratin-A Serve as Potential Inhibitors against Ns2b/Ns3 Protease. Virol Mycol. 11:224.
Received: 07-Jan-2022, Manuscript No. VMID-22-15459; Editor assigned: 10-Jan-2022, Pre QC No. VMID-22-15459 (PQ); Reviewed: 24-Jan-2022, QC No. VMID-22-15459; Revised: 28-Jan-2022, Manuscript No. VMID-22-15459 (R); Published: 04-Feb-2022 , DOI: 10.35841/2161-0517.22.11.224
Copyright: © 2022 Gware P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.