Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 4
Halo blight disease caused due Pseudomonas syringae results extensive losses in dry Beans. Phage therapy can be tried as an alternative treatment. For evaluation of synonymous codon usage and codon variation, 20 phages of Pseudomonas has been taken for testing. The effect of GC concentration in the phages can be considered for analyzing virulence in these phages. Mutational biasing and translational selection are also the important factors for predicting the appropriate biasing, which can be analyzed through Nc plot and correspondence analysis. Our analysis indicating that out of 20 phages 3 phages namely phage D3, D3112 and 119X are extremely virulent as they have high translational efficiency. Based on our data, we conclude that the phage D3 will be best suited as a phage therapy for treatment against halo blight disease.
Keywords: Halo blight; Relative Synonymous Codon Usage; Correspondence analysis; Translational selection; Multivariate statistical analysis; Pseudomonas syringae
RSCU: Relative Synonymous Codon Usage; CA: Correspondence Analysis; Nc; Effective number of codons; GC3s:The frequency of (G + C) at synonymous third codon positions.
Halo blight disease (Saettler et al., 1991) observed through world wide and it can cause extensive losses in dry beans. Pseudomonas syringae is a legume pathogen of worldwide importance and is mainly responsible for Halo blight in Beans (Burkholder, 1926; Burkholder, 1930). Phage therapy of this bacterial pathogen can be tried as an alternative treatment for protecting these legumes against such losses.
Many amino acids are coded by more than one codon and therefore the multiple codons for a given amino acid are synonymous (Ikemura, 1985). However many genes displays a non random usage of specific amino acid and the measure of the extent to this non randomness is given by Relative Synonymous Codon Usage (Sharp et al., 1987). Some genes have extremely biased codon usage: these genes appear to be expressed at higher levels, and other genes (apparently those expressed at low levels) have relatively unbiased codon usage. The other factors which are responsible for variation in codon usage are mutational biasing (Levin et al., 2000) and translational selection (Grantham et al., 1981).
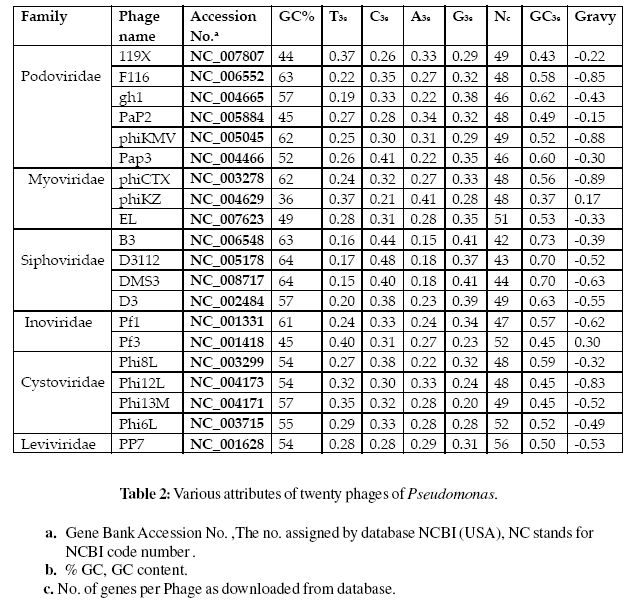
The total of 1214 genes of twenty phages of Pseudomonas are considered which are classified in six families viz. podoviridae, myoviridae, siphoviridae, inoviridae, cystoviridae and leviviridae (Krieg et al.,1984). The genome of Pseudomonas phages shows a considerable variation in their genome size ranging from 2300 – 280000 bps. The Genome of Pseudomonas phage is rich in G-C content which an average accounts for nearly 55% of the total genome content. We found that the optimal codons exhibited variation (AT- or GC-ending codons) in different phages. We also found that genes that were specifically expressed had different patterns of codon usage and local genomic GC (GCg) content. Our efforts to work on this project will provide a path for treatment of halo blight using phage therapy of Pseudomonas. In this paper, we studied the synonymous codon usage bias in all the phages of Pseudomonas whose genomic sequences are known.
The gene sequences of twenty different phages of Pseudomonas were retrieved from NCBI (http:// www.ncbi.nlm.nih.gov/). The gene sequences are retrieved in the FASTA format. The total of 1214 genes is considered for analysis. These genes are of twenty different phages of six families namely podoviridae, myoviridae, siphoviridae, inoviridae, cystoviridae and leviviridae, and were used to study codon bias in these phages. The basic nature and status of the above twenty phages are presented in Table 1.

The gene number for these phages varies from 4 to 301. All the above phage genomes were extracted from featurable table of genome according to gene bank records and all this gene sequences were used for the comparative analysis of the codon usage studies.
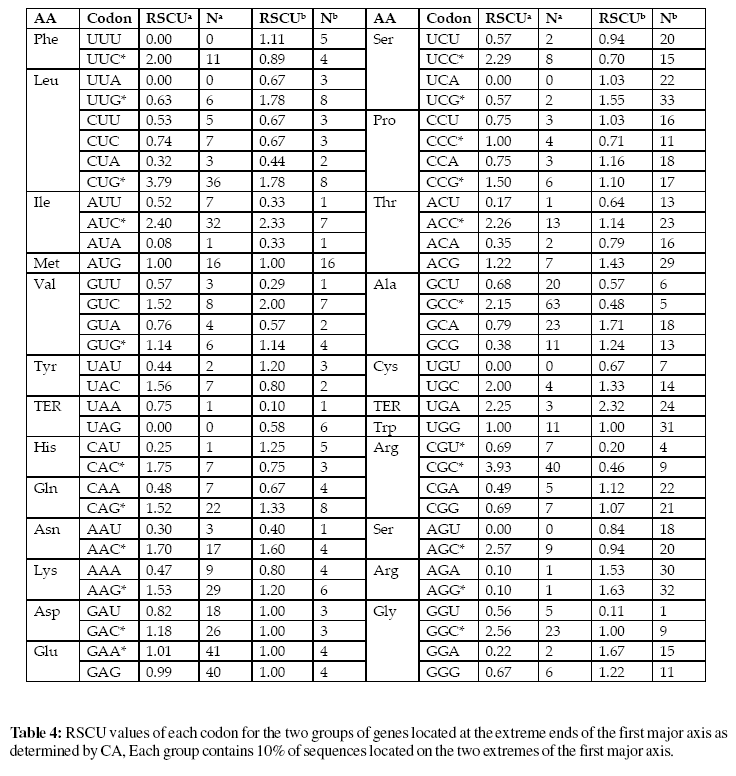
For multivariate analysis we performed Correspondence Analysis (Greenacre, 1984) which is available on CodonW 1.3 . The Relative Synonymous Codon Usage (Sharp et al., 1987) identifies when a codon is being used more frequently than expected and when it is being used less frequently than expected RSCU values are the number of times a particular codon is observed, relative to the number of times that the codon would be observed in the absence of any codon usage bias. Sometimes the observed frequency will be greater than the expected frequency if RSCU value is greater than 1.00, and sometimes it will be less when RSCU value is less than 1.00. RSCU values of each codon for the two groups of genes located at the extreme ends of the first major axis are determined by Correspondence analysis. Each group contains 10% of sequences located on the two extremes of the first major axis.
A3s, T3s, G3s, C3s, are the frequencies of the bases A (adenine), C (cytosine), G (guanine), and T (thiamine) occurring in codon position in the genome and GC3s is the G+C distribution at the synonymous third positions of codons. This G+C content is considered to be the most effective cause of mutational pressure (Sueoka et al. 2000). Nc value is an effective number of codons measure that quantifies how far the codon usage of a gene deposits from equal usage of synonymous codons (Wright, 1990). Nc values range from 61 for a gene that tends to use all codons with equal frequency to 20 for a gene that is effectively using only a single codon for each amino acid.
There is a relationship between Nc and the base composition of a gene with genes that have more biased base compositions being expected to have lower Nc values. Usually, lower Nc values might be dictated by the base composition of the gene. This might be taken as evidence that there is some kind of selective pressure on the gene to use a smaller subset of codons. This selective pressure could be translational selection for ‘optimal’ codons. Optimal codons are those that correspond to the major abundance tRNA for that amino acid. In such circumstances, there could be a selective pressure to use a particular codon that corresponds to this tRNA (Dong et al., 1996).
The copy number of tRNA species in Pseudomonas phage D3 strain and corresponding anticodon sequences were determined by the program tRNA scan-SE . It has been shown that the pattern of codon usage in the highly expressed genes of Escherichia coli and Saccharomyces cerevisiae correlates very strongly with the known abundances of the iso-accepting transfer-RNAs (tRNAs) ) (Ikemura,1981; Bennetzen et al.,1982, Ikemura,1982; Sharp et al.,1991). The advantage of this system (translational selection) is self-evident- using a codon for which there is an abundant cognate tRNA can speed up the process of mRNA translation.
The graph was plotted between various attributes of codon usage like graph of Nc-GC3s, Axis 1 - Axis 2, AXIS 2 - Nc were plotted by the SIGMA PLOT 9.0 . The Correlations coefficient between the positions of genes along the first two major axis with different parameters for codon usage was calculated through SYSTAT 11.0. The alignment among the 20 phages for generating an aligned sequence for dendogram is produced by Clustalw 1.83. A dendrogram representing the extent of divergence in synonymous codon usage among the total phages of pseudomonas was constructed by the DS GENE-SCAN. The tRNA counts present in the genome of phage are calculated by tRNA SCAN SE Server.
Variations in Synonymous Codon Usage
The values of RSCU had been determined in 1214 genes of all the 20 phages of Pseudomonas. It has been observed that all the phages carry GC rich genome. G and C ending codon are predominant in all phages. The concentration of GC present in overall genome of the phage ranges from 36% (phage phiKZ) to 64% (phage DMS3 and phage D3112). To detect codon usage variation if present in any gene of the above-mentioned phages, effective number of codons used by a gene (Nc) and the (G + C) percentage of the synonymous third positions of the codons (GC3s) were determined.
The value of GC3s ranges from 0.144 to 0.912 with an average of 0.549 whereas the value of Nc ranges from 22.74 to 61 with an average of 48.16. The marked intragenomic variation in GC3s (standard variation > 7%) and in Nc values (standard deviation > 4.4% except for phage PP7). These observations indicate that there is a significant heterogeneity in composition within the phage genome of Pseudomonas. The average codon usage bias and the base composition of 20 pseudomonas phage are mentioned in Table 2:
The Effect of Mutational Bias on Codon Usage Variation
A study of correlations between introns and coding region base composition shows that variation in mutation pattern also contributes to codon bias variation (Kliman et al., 2003). The strength of base composition correlations between introns and codon third positions is greater for genes with low codon bias than for genes with high codon bias. One direct effect of mutation bias on genome evolution is to influence genome composition, which can be measured by G+C content. For analyzing the determinants of codon usage bias in the phages of Pseudomonas, Nc plots (a plot of Nc versus GC3s) and the correspondence analysis (CA) are used widely.
The Nc plot drawn for the genes of 20 Pseudomonas phages are displayed in Figure 1. Some of the points especially of phage phiKZ, phage119X and phage B3 lie on the expected curve towards GC-poor regions (GC value 0.144 to 0.2) which certainly originates from extremely mutational bias. It is evident from the figure that a considerable number of genes lie well below the expected curve, indicating that codon usage bias of these genes are influenced by the forces other than genomic GC composition. Points demonstrated by phage D3 and phage D3112 lie away from the expected curve in comparison with the rest of the phage genes which indicate that the effect of mutational bias on codon usage variation in the former three phage genes is very weak. This phenomenon was further verified with other statistical analysis like correspondence analysis.

Figure 1: Nc plot of genes from 20 Pseudomonas phages.
The correspondence analysis of RSCU values of 1214 genes of the 20 Pseudomonas phages confirms that mutational bias and other factors are also responsible for codon usage variation. The main objective to plot genes in axis 1 and axis 2 space is finding of optimal codons. Optimal codons are defined as those codons that occur significantly more often in highly expressed genes relative to their frequency in lowly expressed genes. Significance is assessed by a twoway chi square contingency test with the criterion of p < 0.01. The advantage of using a test of significance to identify optimal codons is that variation in codon usage between highly and lowly expressed genes, that is due to random noise is suppressed. Correspondence analysis is a multivariate statistical analysis technique to study codon usage variation among genes (Wright, 1981). In this analysis, the data are plotted in a multidimensional space of 59 axes (excluding Met, Trp and stop codons), then the most prominent axes are determined that contribute to the codon usage variation among the genes.
The positions of genes along the first as well as the second major axis (generated by CA) are analyzed with the nucleotide composition at the third codon shows that the first major axis is positively correlated with G3 (r, correlation coefficient = 0.067), C3 (r = 0.261 with P < 0.05) and negatively correlated with A3 (r, correlation coefficient = - 0.043) and T3 (r correlation coefficient = -0.104). In contrast the reverse is true for the second major axis.
The correlation coefficient between the second axis and GC3s is relatively small as compared to that between the axis1 and GC3s (Table 2). But it is worth mentioning that the axis2 exhibits strong negative correlation with G3s and positive correlation with C3s (Table 3). These observations indicate that G3s and C3s interact synergistically in the first principal axis resulting in the increase of GC3s content, but antagonistically in the second principal axis so that increase in the frequency of C3s is accompanied by a decrease in G3s and vice-versa.

The position of genes of the first two major axes (Figure 2) clearly shows that the majority of genes of phage D3, phage D3112 and phage 119X are not clustered with genes of the other phages. To investigate the difference between these two clusters of genes, the codon usage of 10% of the genes located at extreme right side of axis 1 was compared with 10% of the genes located at the extreme left side of axis 1. To access the variation in codon usage between these two genes, chi-square tests were performed taking P < 0.01 as the significant criterion. The number and occurrence of each codon and its RSCU values for the two groups of genes are displayed in Table 3.

Figure 2: Position of genes from 20 Pseudomonas phages along the two major axes of variation in the correspondence analysis on RSCU values.
Out of 21 predominant codons there are 11 C ending codons and 8 G ending codons which actually represent 90.47% of total G and C ending codons. This result suggests that genomic GC composition has a profound effect on in separating the genes along the first major axis according to their RSCU values. It has been reported that RNY codons are more advantageous for translation (Shepherd, 1981). It was also demonstrated that in highly expressed genes of Escherichia coli, C is the prominent base at the third codon position (Gutiérrez et al., 1996). The high occurrence of C ending codons in highly expressed genes demonstrates that compositional constraints are not the only factor in determining the codon usage variation in this organism. If compositional constraints are the only dictator in codon usage variation in this organism, the base composition in the third codon position among these optimal codons should have also A or T ending codons as observed in the overall RSCU values of this organism. A similar type of observation was also reported for Plasmodium falciparum (Musto et.al., 1999).
Cluster analysis has been successfully used to study the frequency of codon usage divergence among the genes of an organism and also among the organisms (Sharp et al., 1986). The codon frequency of 64 codons for each organism was compared with the codon frequency with all other organisms. Figure 4 shows the clustering produced by UPGMA (unweighted pair group method using arithmetic averages) method (Sneath et al., 1973). From the Figure 4 it is evident that there are two distinct branches for the twenty Pseudomonas phages. Second branch out of the two branches comprises of PP7, PaP3, phiKMV, gh-1, phi-12L, phi-6l and phiEL and the rest are in first branch. Phage D3, D3112 and 119X are of same branch and it suggest that they have nearly similar synonymous codon usage pattern, provided 119x is of family podoviradae, while the other two phage D3 and D3112 are from Siphoviridae.

Figure 3: Plot between the average value of Axis 1 and Axis 2 of 20 phages of Pseudomonas syringae.

Figure 4: A phylogram representing the extent of divergence in synonymous codon usage of 20 phages of Pseudomonas syringae.
The above cluster analysis has not only supported our correspondence as well as variance analyses (mentioned above) but also assisted in understanding the intra- and inter- genomic diversities of the Pseudomonas bacteriophages in a much better way.
The Influence of Translational Selection Over Codon Usage Variation in Pseudomonas Syringae
In Caenorhabditis elegans and Drosophila melanogaster, which are characterized by extensive variation in codon usage, the factors governing the choices have been attributed to equilibrium between mutational biases and translational selction (Shields et al.,1981 ; Sharp et al, 1989; Moriyama et al.,1992; Carulli et al.,1993; Akashi, 1993; Akashi, 1997; Stenico et al.,1994 ; Moriyama et al., 1997; Powell et al., 1997). In many organisms, selection acts on synonymous codons to improve translation. The selection on synonymous codon use in E. coli is largely due to selection for translation accuracy (Gouy, 1982). The plot between second major Axis and Nc values suggest that a substantial number of phage genes, particularly to the phages of D3, D3112 and 119X have lower Nc values as compare to other phage genes (Figure 5).

Figure 5: Plot between the genes of 20 Pseudomonas phages and Nc values.
The first major axis is negatively correlated with Nc, whereas the second major axis is positively correlated with Nc (Table 3). This suggests that considerable number of phage genes carrying GC-rich codons have low Nc values. On the basis of these results, we urge that a balance between mutation and selection due to translational efficiency is strongly operating in selecting the codon usage variation among the genes of Pseudomonas phages. Besides phages D3, D3112, 119X mostly carry highly expressed genes.
Various reports suggests that the synonymous codon choices appear to be positively correlated with the relative abundance of tRNAs, with the correlation being very strong for highly expressed genes (Ikemura,1981; Bennetzen et al.,1982; Ikemura,1982; Ikemura,1981; Bennetzen et al., 1982; Gouy et al.,1982; Sharp et al., 1986; Bulmer, 1988; Bulmer, 1991; Kanaya et al.,1991)In several organisms, cellular tRNA abundance was shown to be directly proportional to their tRNA copy number (Kanaya et al.,1991; Kanaya et al.,2001) Table 4.
To point any positive correlation between host tRNA abundance and synonymous codon usage of phage D3 and phage D3112 a comparative analysis was made between the copy numbers of Pseudomonas syringae specific tRNA and over represented synonymous codons of phage D3 and phage D3112 separately (Table 5).
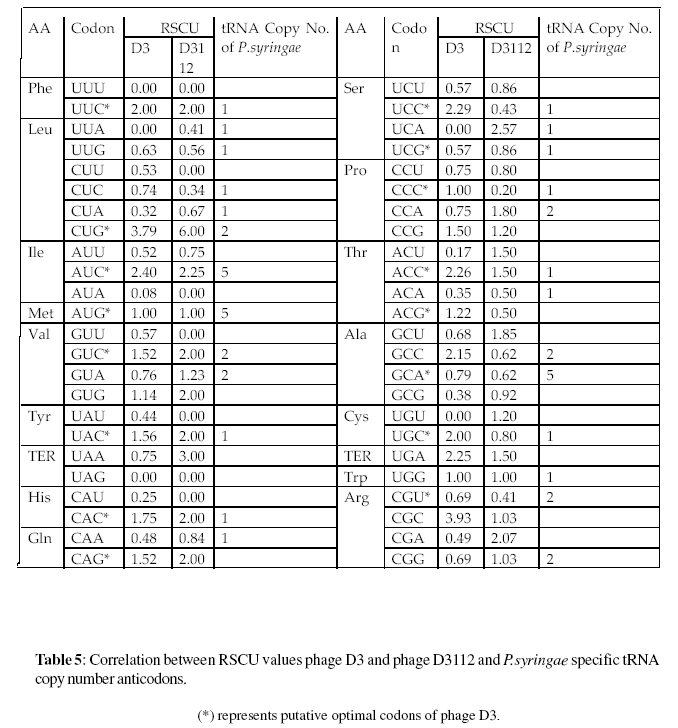
It was found that out of total 23 over-represented codons of D3, 20 are recognized by those Pseudomonas syringae specific tRNAs that have at least one copy in the cell. In contrast, 2 less over-represented codons of D3112 are recognized by abundant tRNAs Pseudomonas syringae specific tRNAs. The data indicates that most of the genes of D3 and D3112 have high translation efficiency.
The study of Synonymous codon usage of 20 phages of Pseudomonas syringae has been done in our work. Out of these 20 phages, phage D3, D3112 and 119X codon usage, their correlation with expression levels and their translational selection clearly suggest that these may be useful for curing Pseudomonas infections.
Further, a comparative analysis of codon usage and RSCU value of the protein coding genes of these 20 phages was done. The cluster analysis (Figure 2 and Table 3) also indicates the similarity in the synonymous codon usage and the divergence among the phages D3, D3112 and 119X. The RSCU value suggest that D3 and D3112 have high GC content and the phage 119X though have high AT rich regions, but the codon variation is uniform in phage 119X (shown in Figure 1). It is observed that D3 carries most highly expressed genes compared with the other phages. Also the over-represented codons of D3 are preferentially reorganized compared with other phages. Based on this data (Figure 5 and Table 5), we suggest that the genes of D3 are expressed rapidly by host’s translation machinery. Several lytic phages had been used successfully to cure infected patients (Sulakvelidze et al.,2001; Markoishvili et al.,2002; Jikia et al.,2005). It is speculated whether to use phages as a mixture of relatively weak and virulent phages or as one of a kind. A comparative analysis on codon usage of E. coli phages T1, T3, T4, T5 and T7 indicates that T4 carries the highest percentage of highly expressed genes among 5 phages and T4-like phages showed immense potential in therapy (Chibani-Chennoufi et al., 2004).
Our results interprets that the codon usage of different phages of Pseudomonas vary significantly which can be suggested due to the occurrence of several factors such as mutational biasing, translational selection etc.
On the basis of our data we suggest that out of twenty phages of Pseudomonas phage D3 will be best suited for the treatment of Pseudomonas infections. Thus the phage D3 may be recommended to the phage therapy for the treatment of halo blight disease of Pseudomonas syringae.
The authors are thankful to the Sub-Distributed Information Center (BTISnet SubDIC), Department of Biotechnology (No. BT/BI/04/065/04), New Delhi, India for financial support.