Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2014) Volume 5, Issue 1
Studies of our experiment are related to the binding of enzymes to antibodies. This involves the formation of a stable, covalent linkage between an enzyme & antibody. We used different Enzymes for this studies which are altered in binding action. Enzymes we used-e.g. Horseradish peroxidase (HRPO), urease, or alkaline phosphatase. Studies of Enzyme and an antigen-specific monoclonal or polyclonal antibody in which neither the antigen-combining site nor the active site of the enzyme is functionally altered. The enzyme most commonly used in the immunoreagent (the antibody enzyme conjugate) preparation is horseradish peroxides. This enzyme is cheap and can be attached to the immunoreagent by a variety of methods. Moreover many chromogenic substrates for it are also available. Our Experiment studies on conjugation of Horseradish Peroxidase (HRPO) to antibody (anti human IgG) will be carried out by Periodate oxidation method. We have prepared Anti human IgG-HRP conjugate in our laboratory. This will be used for ELISA and western blot experiments and IgG purified from different human serum samples would be used as the antigen in the above experiments. This experiment is carried out in two different steps mentioned as: Preparations of anti-human IgG-Horseradish peroxidase conjugate, Characterization of isolated human IgG.
Keywords: HRPO; Immunoglobulin’s; Enzyme conjugation; Blotting techniques; Periodate oxidation method; Binding of enzymes to antibodies
Conjugation of enzymes to antibodies
Conjugation of enzymes to antibodies involves the formation of a stable, covalent linkage between an enzyme [e.g. Horseradish Peroxidase (HRPO), urease, or alkaline phosphatase] and an antigen-specific monoclonal or polyclonal antibody in which neither the antigen-combining site nor the active site of the enzyme is functionally altered. The chemistry of cross-linking HRPO or urease to immunoaffinity purified monoclonal or polyclonal antibodies (IgG) is presented in the chemistry of cross-linking alkaline phosphatase to antibodies is presented in Figure 1.

Figure 1a: (Precipitation reactions description: (a) Polyclonal antibodies can form lattices, or large aggregates, that precipitate out of solution. However, if each antigen molecule contains only a single epitope recognized by a given monoclonal antibody, the antibody can link only two molecules of antigen and no precipitate is formed. (b) A precipitation cur ve for a system of one antigen and its antibodies. This plot of the amount of antibody precipitated versus increasing antigen concentrations (at constant total antibody) reveals three zones: a zone of antibody excess, in which precipitation is inhibited and antibody not bound to antigen can be detected in the supernatant; an equivalence zone of maximal precipitation in which antibody and antigen form large insoluble complexes and neither antibody nor antigen can be detected in the supernatant; and zone of antigen excess in which precipitation is inhibited and antigen not bound to antibody can be detected in the supernatant).

Figure 1b: (Reaction Description: Conjugation of Horseradish Peroxidase (HRPO) to antibody (IgG) using the periodate oxidation method. The method involves three chemical steps: (1) sodium periodate (NaIO4) oxidation of the carbohydrate side chains of HRPO, (2) Schiff base formation between activated peroxidase and amino groups of the antibody, and (3) sodium borohydride (NaBH4) reduction of the Schiff base to form a stable conjugate. Contributed by Scott E. Winston, Steven A. Fuller, Michael J. Evelegh, and John G.R. Hurrell Current Protocols in Molecular Biology (2000) 11.1.1-11.1.).
The enzyme most commonly used in the immunoreagent (the antibody enzyme conjugate) preparation is horseradish peroxides. This enzyme is cheap and can be attached to the immunoreagent by a variety of methods. Moreover many chromogenic substrates for it are also available [1,2].
Background information
The conjugation of HRPO to antibody is dependent on the generation of aldehyde groups by periodate oxidation of the carbohydrate moieties on HRPO. Combination of these active aldehydes with amino groups on the antibody forms Schiff base upon reduction by sodium borohydride, become stable. For urease conjugation, crosslinking the enzyme and antibody with MBS (m-maleimidobenzoyl N-hydroxysuccinimide ester) is achieved through benzoylation of free amino groups on antibody. This is followed by thiolation of the maleimide moiety of MBS by the cysteine sulfhydryl groups of urease. The advantages of urease conjugates are their stability in solution at normal working dilutions, the rapid turnover rate of the enzyme, the easily discernible color change when substrate is added, and the fact that urease is not found in most mammalian or bacterial systems. The disadvantage is that since no precipitable substrate is available, urease conjugates cannot be used for immunohistology or western blotting. Alkaline phosphatase conjugates are useful for all types of immunological assays depending on the alkaline phosphatase substrate used (i.e., p-nitrophenyl phosphate in diethanolamine is the preferred substrate for ELISA with colorimetric detection, 4-methylumbelliferyl phosphate is useful for ELISA with fluorimetric detection, and nitrobluetetrazolium/5-bromo-4-chloro-3-indolyl phosphate is the preferred substrate for western blotting). Alkaline phosphatase conjugates are as stable as urease conjugates and more stable than HRPO conjugates. Endogenous phosphatases can cause false positive reactions. However, levamisole will inhibit alkaline phosphatase in many mammalian tissues but not the alkaline phosphatase (i.e., bovine intestinal) used in the conjugates and for this reason levamisole may be added to the substrate solution. The one-step glutaraldehyde method is the simplest available procedure for preparing alkaline phosphatase– antibody conjugates. Various alternative procedures for preparing alkaline phosphatase conjugates have been compared. The sensitivity that can be achieved with HRPO, urease, or alkaline phosphatase conjugates is comparable and between 1 ng/ml and 10 ng/ml of antigen can be detected [3,4].
Critical parameters
The most critical parameters of both conjugation methods are the quality of enzyme and the cross-linking reagents. These reagents should be tested as described in the protocol before conjugating to larger quantities of antibodies. It is imperative that the m-maleimidobenzoyl N-hydroxysuccinimide ester (MBS), sodium periodate (NaIO4) and sodium borohydride (NaBH4) be stored in a desiccator and that solutions containing these chemicals be prepared immediately prior to use. The method described is applicable to most antibodies and should produce conjugates that are useful for developing an ELISA for detecting sensitively and specifically for a given antigen. However, not all antibodies conjugate in an identical manner. It may be necessary to vary the ratio of MBS/antibody or urease/antibody for the urease conjugation and the NaIO4/HRPO and HRPO/antibody ratios for a given HRPO conjugation. The quality and grade of alkaline phosphatase is crucial to the generation of effective conjugates. Immunoassay grade material is recommended over lower grades, and the enzyme should not be conjugated beyond its expiration date. In the case of polyclonal antisera, the specificity and titer of the antiserum will be reflected in the conjugate and any purification procedures that increase these values, such as immunoaffinity chromatography will enhance conjugate performance. The selection of an optimal conjugation time for preparing alkaline phosphatase–antibody conjugates varies for different antibodies, in particular when monoclonal antibodies are used. In contrast, polyclonal antibodies may be reliably conjugated in 120 min.
Trouble shooting
There are several factors that may contribute to the production of poor enzyme-antibody conjugates. It is important to determine first whether a poor conjugate is the result of inactivation of either the antibody or the enzyme (or both) or the result of insufficient or excessive cross-linking. The affinity of the antibody for substrate can be measured by determining the presence of bound antibody with another immunoassay employing anti-antibody conjugated to a different enzyme. Enzyme activity can be measured by cleavage of substrate at different enzyme concentrations. Precipitation of material in the conjugate solution or opaque solutions, are indicative of excessive crosslinking. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis is useful for monitoring the extent of cross-linking by determining the Mr of the cross-linked species. Insufficient cross-linking usually results from the use of inactive or poor-quality crosslinking agents. Excessive cross-linking and inactivation of antibody or enzyme can be eliminated by either reducing the concentration of antibody and enzyme or by reducing the time of reaction. It may not be possible to generate effective alkaline phosphatase conjugates with all antibodies using the one-step glutaraldehyde method. An alternative is to try a different conjugation technique. Another alternative is to use an antispecies antibody–alkaline phosphatase conjugate to detect the antibody in question. These reagents may be purchased or prepared using the above technique.
Anticipated results
The yield and titer of the resultant conjugate will depend on the original antibody’s properties and specific application. It is difficult to estimate the yield or working dilution of the conjugates, as it is dependent on numerous factors such as antibody affinity, type of ELISA, and quality of antigen. In general, the working dilutions range from 1:100 to 1:10,000.
In our studies conjugation of horseradish peroxidase (HRPO) to antibody (anti human IgG) will be carried out by periodate oxidation method. Anti-human IgG-HRP conjugate prepared in our laboratory. This will be used for ELISA and western blot experiments (IgG purified from different human serum samples would be used as the antigen in the above experiments). Thesis work will be carried out in two parts for the sake of clarity and convenience.
Preparations of anti-human IgG-HRP conjugate
Prepared anti human IgG HRP conjugate will be used for the following experiments
Qualitative ELISA for detection of human IgG
Identification of IgG by western blotting
Toward this objective, IgG from different human serum samples will be isolated and it will be used for the above experiments.
Characterization of isolated human IgG
1. Estimation of protein content
2. SDS-PAGE analysis: Determination of purity, Determination of molecular weight
Preparations of anti-human IgG-HRP conjugate
Chemicals and reagents: Antibody labeling teaching kit was purchased from Bangalore Genie Pvt Ltd, Bangalore India.
Materials provided in the kit are given below:
1. Antigen spotted strip (Human IgG spotted)
2. Antibody for coupling (Anti human IgG)
3. Carbonate buffer
4. Desalting column G 25 (2 ml)
5. Oxidation tubes
6. Reductant solution
7. Stabilizer
8. 10 X ELISA buffer
9. 10 X Phosphate buffer saline (PBS)
10. 10 X TMB/H2O2
Other requirements
1. Micropipettes and tips
2. Measuring cylinder, test tubes and beakers
3. 2,5 and 10 ml pipettes
4. Burettestand
5. Distilled water.
Oxidation of HRP
0.2 ml of distilled water was added to the oxidation tube containing HRP and sodium Meta-periodate. The solution was stirred for 20 min at room temperature. (Note: colour changes from orange to green).
Coupling
0.2 ml of carbonate buffer was added to antibody coupling tube and the contents were mixed thoroughly. Immediately after mixing, oxidized HRP was added to the antibody solution and the contents were stirred for 2 h at room temperature. After 2 h, 10 μl of reductant solution (borohydride) was added and the solution was mixed for 10 min at room temperature (Figure 2).

Figure 2: The most commonly used method for labeling IgG molecules with HRP exploits the glycoprotein nature of the enzyme. The saccharide residues of HRP are oxidized with sodium meta-periodate to produce aldehyde groups that can react with the amino groups of the IgG molecule and the Schiff’s bases formed are then reduced by borohydride to give a stable conjugate.
Desalting
The desalting column was first equilibrated with 20 ml of 1X PBS. After allowing the reaction mix to settle down the supernatant was loaded on to the desalting column that was previously equilibrated with PBS. After running down the reaction mixture into the column 0.1 ml of 1X PBS was added along the walls of the column to wash down the reaction mixture sticking onto the column. Once the PBS entered the bed of the column, 1 ml of 1X PBS, was added at a time till the entire colored fraction (HRP conjugate) eluted out of the column completely (Figure 3) Immediately after collecting the HRP conjugate 0.5 ml of stabilizer was added to the HRP conjugate and the conjugate was stored at 4°C until use. After eluting the HRP conjugate, the column was washed with 5 ml of 1X PBS and stored at 4°C.

Figure 3: Desalting of HRP-conjugate using a gel filtration column to remove the borohydride.
Titration
The HRP conjugate was diluted with 1X assay buffer. Four antigen (Fraction no. 2 from serum sample 1, 2 and 3) spotted nitrocellulose strips were labeled 1:1000, 1:2000, 1:4000 and 1:8000 and then the strips were agitated in appropriately diluted conjugate (i.e. 1:1000, 1:2000, 1:4000 and 1:8000 diluted anti human IgG HRP conjugate (Figure 4).

Figure 4: 1:1000, 1:2000, 1:4000 and 1:8000 diluted anti human IgG HRP conjugate.
The strips were then incubated at room temperature for 30 min with constant shaking. After incubation, the strips were washed 5 times with 2-3 ml of rinse buffer (1 min each wash). After each wash the old buffer was replaced with fresh buffer. After the fifth wash the strips were washed with distilled water once. After washing, the strips were agitated in 2 ml of substrate solution (TMB/H2O2) till blue/ grey colored spots were observed. Soon after the spots developed the strips were removed before the background turned dark. The highest dilution at which the spot are seen is the titer value of the conjugate [6-8].
Chemicals and reagents
10N sodium hydroxide
a. 4 g sodium hydroxide
b. 10 ml of distilled water
4 g sodium hydroxide was dissolved in about 8 ml of distilled water and then the volume was made up to 10 ml with distilled water.
Phosphate buffer saline (PBS) pH 7.4 (equilibration buffer):(20x/1000 ml)
a. Sodium chloride=180 g
b. Potassium dihydrogen orthophosphate=2.88 g
c. Disodium hydrogen ortho phosphate 2H2O=17.78 g
d. 1000 ml of distilled water
180 g sodium chloride, 2.88 g of dipotassium hydrogen orthophosphate and 17.78 g of disodium hydrogen orthophosphate was dissolved in about 800 ml of distilled water. After adjusting the pH to 7.4 with 10 N sodium hydroxide, the volume was made up to 1000 ml with distilled water (2.1 ml 0f 10N NaOH was required to adjust the pH to 7.4). To prepare 1X buffer, the 20X buffer was diluted 20 times (e.g. 50 ml of 20 X PBS was diluted to 1000 ml with distilled water (+950 ml of distilled water).
Elution buffer: (0.1M citrate buffer pH=4.6)
0.1 M citric acid=2.1 g
2.1 g citric acid was dissolved in about 80 ml of distilled water and then the volume was made up to 100 ml with distilled water.
0.1 M sodium citrate=2.94 g
2.94 g sodium citrate was dissolved in about 80 ml of distilled and then the volume was made up to 100 ml with distilled water.
To prepare 0.1 M citrate buffer pH 4.6, 25.5 ml of solution (a) was mixed with 24.5 ml of solution (b) and then the volume was made up to 100 ml with distilled water.
Neutralization buffer: (2 M Tris (hydroxy methyl) amino methane)
Tris (hydroxy methyl) amino methane=2.42 g, 10 ml of distilled water
2.42 g of Tris (hydroxy methyl) amino methane was dissolved in about 8 ml of distilled water and then the volume was made up to 10 ml with distilled water.
Storage buffer: (equilibration buffer containing 0.05% (w/v) sodium azide)
25 mg sodium azide, 50 ml of equilibration buffer, 25 mg of sodium azide was dissolved in 50 ml of equilibration buffer.
Protein A affinity column: (column size-2 ml): Protein-A affinity column was purchased from Bangalore Genie Pvt Ltd Bangalore India.
Serum samples: Human serum samples 1, 2 and 3.
Other requirements
1. Test tubes and test tube stand
2. 2, 5 and 10 ml glass pipettes
3. Glass beakers and conical flasks
4. 10, 50 and 100 ml measuring cylinder
5. Micro pipettes and tips; 2-20, 20-200 and 100-1000 μl
6. Eppendorf tubes (1.5 ml)
7. Refrigerated centrifuge
8. Burettestand
9. Tissue paper
10. Distilled water
11. 1ml quartz cuvettes
12. UV-VIS spectrophotometer
13. Graph sheet, pencil and eraser
Sample preparation
Serum sample was mixed with equilibration buffer in 1:1 ratio in an eppendorftube. After mixing, the sample was subjected to centrifugation at 6000 rpm at 4°C for 10 min to remove particulate matter.
Protein A Column Chromatography (Bed volume 2 ml)
After the column was brought to ambient temperature, the column was equilibrated with 10 bed volumes of equilibration buffer. After equilibration, 0.9 ml of the 1:1 diluted clear serum sample was loaded on to the column. While loading, the flow rate of the column was adjusted to 1 ml/5 min. Once the serum sample passed through the column, the column was washed with equilibration buffer till the A280 of the equilibration buffer was less than 0.1 O.D. (About 10 bed volumes of equilibration buffer was required for washing). After the unbound materials were removed by washing, the bound proteins were eluted with 15 bed volumes of elution buffer. 1ml fractions were collected in tubes containing neutralizing buffer (25 μl/ml) (Figure 5).

Figure 5: A direct ELISA was performed with serum samples 1, 2, 3, fraction no. 2 from serum samples 1, 2 and 3. Individual antigens were coated in to micro titer ELISA plates at a concentration of 10 μg/ml in 0.1M sodium carbonate and bicarbonate buffer. After coating, wells were washed with rinse buffer. Unadsorbed sites were blocked with blocking buffer .Excess blocking buffer was removed by washing with rinse buffer. After blocking, the wells were treated with anti-human IgG -HRP conjugate. Excess labeled antibody was removed by washing the wells with rinse buffer and finally TMB/H2O2 was added in to each well. As negative controls rabbit IgG were coated in few wells. Results of such an experiment are shown in the figure.
After collecting the fractions, A280 for each fraction was measured at 280 nm in a UV-VIS spectrophotometer. Serum samples 2 and 3 were treated similarly. After taking the absorbance value a graph was plotted with fraction number on x-axis vs. absorbance values on y-axis.
Chemicals and reagents
Antigen:
a. Pure human IgG was purchased from Bangalore Genie Pvt Ltd, Bangalore India.
b. Pure Rabbit IgG was purchased from Bangalore Genie Pvt Ltd, Bangalore India.
c. Serum samples 1, 2, 3
d. Fraction no 2 ( serum samples 1, 2 and 3)
Samples were prepared at a concentration of 5μg/ml individually in coating buffer (0.1M sodium carbonate and bicarbonate buffer pH 9.6).
Antigen coating buffer: (0.1 M sodium carbonate bicarbonate buffer pH 9.6)
0.1 M sodium carbonate=1.06 g/100 ml: 1.06 g of sodium carbonate was dissolved in about 80 ml of distilled water and then the volume was made up to 100 ml with distilled water.
0.1M sodium bicarbonate=1.26 g/150 ml:
1.26 g of sodium bicarbonate was dissolved in about 80 ml of distilled water and then the volume was made up to 150 ml with distilled water.
To prepare 0.1 M sodium carbonate bicarbonate buffer pH 9.6, 84 ml of (a) was added to 150 ml of (b) and mixed.
Phosphate buffer saline (PBS) pH 7.4 (equilibration buffer): (20x/1000 ml)
a. Sodium chloride=180 g
b. Potassium dihydrogen orthophosphate=2.88 g
c. Disodium hydrogen orthophosphate 2H2O=17.78 g
d. 1000 ml of distilled water
180 g sodium chloride, 2.88 g of dipotassium hydrogen orthophosphate and 17.78 g of disodium hydrogen ortho phosphate was dissolved in about 800 ml of distilled water. After adjusting the pH to 7.4 with 10N sodium hydroxide, the volume was made up to 1000 ml with distilled water. (2.1 ml of 10 N NaOH was required to adjust the pH to 7.4). To prepare 1x buffer, the 20X buffer was diluted 20 times (e.g. 50 ml of 20X PBS was diluted to 1000 ml with distilled water (+950 ml of distilled water).
Rinse buffer: 50 ml of 20X PBS was diluted to 1000 ml with distilled water. After mixing, 1 ml of tween 20 was added to it.
Serum diluent buffer: 1 g of egg albumin was dissolved in 100 ml of rinse buffer.
Labeled antibody: Anti-human IgG HRP conjugate prepared in our laboratory was used.
Substrate solution: Tetramethylbenzidine/hydrogen peroxide (TMB/H2O2):
TMB/H2O2 was purchased from Bangalore Genie Pvt Ltd, Bangalore India. TMB is a noncarcinogenic chromogen used in assays involving Horseradish Peroxidase (HRP) enzyme. TMB/H2O2 for ELISA produces a soluble blue colored product with HRP. The reagent is supplied as 20X concentration. Just before use 1 volume of the substrate solution was mixed with 19 volumes of distilled water.
Other requirements
1. 96 well micro titer plates.
2. Micro pipettes 2-20 and 200-1000 μl and tips.
3. Glass pipettes 5 ml and 10 ml.
4. Glass beakers and conical flasks
5. Measuring cylinders
6. Wash bottle
7. Incubator set at 37°C.
8. Moist chamber (plastic box with a layer of wet cotton)
9. ELISA Reader.
Method
All assays were carried out in 96 well flat bottomed polystyrene plates.
Antigen coating: 100 μl of antigen at a concentration of 5 μg/ml in 0.1 M sodium carbonate–bicarbonate buffer, pH 9.6 was coated onto each well and the plate was incubated for 30 min at 37°C (Few wells coated with rabbit IgG was used as negative control). After incubation, excess antigen was removed from the well by washing 5 times with rinse buffer (PBS with 0.1% tween 20).
Blocking non specific sites: After washing, non-specific sites were blocked by addition of 300 μl serum diluent buffer (SDB, containing PBS with 0.1% tween-20 and 1% egg albumin) into each well and plate was incubated for 30 min at 37°C. After incubation, plate was washed 5 times with rinse buffer.
Labeled antibody (Anti-human IgG HRP-conjugate and goat anti-human IgG HRP conjugate): After washing, 100 μl of 1:2000 diluted anti-human IgG-HRP conjugate was added to each well and the plate was incubated for 30 min at 37°C. Excess antibody enzyme conjugate was removed by washing the well 5 times with rinse buffer.
Substrate solution: After washing, 100 μl of the chromogenic substrate (TMB/H2O2) was added to each well and the plate was incubated for 10 min at room temperature till colour developed. Colour developed was read at 450 nm in a micro titter plate auto reader ELISA reader (Figure 6).

Figure 6: In Western blotting, a protein mixture is (a) treated with SDS, a strong denaturing detergent, (b) then separated by electrophoresis in an SDS polyacrylamide gel (SDS-PAGE) which separates the components according to their molecular weight; lower molecular weight components migrate farther than higher molecular weight ones. (c) The gel is removed from the apparatus and applied to a protein-binding sheet of nitrocellulose or nylon and the proteins in the gel are transferred to the sheet by the passage of an electric current. (d) Addition of enzyme-linked antibodies detects the antigen of interest, and (e) the position of the antibodies is visualized by means of a reaction that generates a highly colored insoluble product that is deposited at the site of the reaction.
Chemicals and reagents
Blotting buffer:
a. 2.42 g of tris base
b. 10.25 g of glycine
c. 200 ml of 20% methanol
d. 800 ml distilled water
2.42 g of tris base, 10.25 g of glycine was dissolved in about 700 ml of distilled water and then 200 ml of 20% methanol was added to it and then the volume was made up to 1000 ml with distilled water.
Phosphate buffer saline (PBS) pH 7.4 (equilibration buffer): (20x/1000 ml)
a. Sodium chloride=180 g
b. Potassium dihydrogen orthophosphate=2.88 g
c. Disodium hydrogen orthophosphate 2H2O=17.78 g
d. 1000 ml of distilled water
180 g sodium chloride, 2.88 g of dipotassium hydrogen ortho phosphate and 17.78 g of disodium hydrogen ortho phosphate was dissolved in about 800 ml of distilled water. After adjusting the pH to 7.4 with 10N sodium hydroxide, the volume was made up to 1000 ml with distilled water (2.1 ml of 10N NaOH was required to adjust the pH to 7.4). To prepare 1x buffer, the 20x buffer was diluted 20 times (e.g. 50 ml of 20 X PBS was diluted to 1000 ml with distilled water (+950 ml of distilled water).
Rinse buffer: 50 ml of 20X PBS was diluted to 1000 ml with distilled water. After mixing;
1 ml of tween 20 was added to it.
Serum diluent buffer: 1 g of egg albumin was dissolved in 100 ml of rinse buffer.
Labeled antibodies: Anti-human IgG HRP conjugate was prepared in our laboratory.
Substrate solution: Tetramethylbenzidine/hydrogen peroxide (TMB/H2O2) was purchased from Bangalore Genie India. TMB is a noncarcinogenic chromogen used in assays involving Horseradish Peroxidase (HRP) enzyme. The reagent is supplied as 20 X concentration. Just before use 1 volume of the substrate solution was mixed with 19 volumes of distilled water.
Fraction no 2 from serum samples 1, 2 and 3: Fraction no 2 from serum samples 1, 2 and 3 were spotted on nitrocellulose membrane strip. Unadsorbed (non-specific) sites on the membrane were blocked by agitating the membrane in 10 ml blocking buffer. After washing, the membrane was agitated in 1:200 diluted goat anti human IgG-FITC. Excess goat anti human IgG-FITC conjugate was removed by washing. After air drying, the membrane was placed in a UV cabinet to observe the fluorescence spot developed.
Other requirements
1. Nitro cellulose membrane (pore size 0.22-0.45 micro meter)
2. Glass pipettes
3. Beakers
4. Conical flask
5. Petri dish
6. Scalpel blade
7. Glass tray or Plastic tray
8. Glass plate platform 25x20 cm
9. Tissue paper rolls
10. Whatman filter paper
11. cissors
12. Forceps
13. Gloves
14. Small electrophoresis unit
15. Electrotransfer unit or small electro blotting system
16. Power supply unit
17. Connecting cords
18. Shaker.
Method
Western blotting is essentially a combination of three techniques electrophoresis (PAGE), blotting (protein blotting) and immunochemical detection (blot development).
Stage I: Separation of proteins on SDS-PAGE
The proteins to be analyzed (fraction no 2 from serum samples 1, 2 and 3) by western blotting was first subjected to separation on a 10- 12% separating gel and 6% stacking gel. Samples were electrophoresed at 80-100 volts.
Stage II: Electro transfer of the separated protein onto the nitro cellulose membrane
Blotting is the transfer of resolved proteins from the gel to the surface of a suitable membrane. The separated proteins are transferred out of the gel either by the capillary action of the buffer or in an electric field (known as electro blotting).
After separation of proteins by SDS-PAGE, the stacking gel was discarded and the required portion of the gel was cut with a scalpel blade, if the whole gel was not to be blotted. Similarly, the nitro cellulose membrane (one layer) and the filter paper (six layers) were cut to the exact size of the gel. The gel, the NC membrane and the filter papers were then soaked in a tray containing the blotting buffer for 10 min. After 10 min, the blotting sandwich was assembled. During sandwich assembly preparation care was taken to avoid air bubbles between the gel and NC membrane. After assembling the blotting sandwich, the cassette was inserted into the apparatus filled with the blotting buffer. The gel was placed towards the cathodic end; since proteins are negatively charged (due to presence of SDS) their migration will be towards anode. After inserting the cassette into the apparatus lid was placed over the buffer tank and with the help of connecting cords the apparatus was connected to power supply and electro transfer was carried out overnight at 50 volts. The presence of SDS facilitates the migration of proteins. Once out of the gel, the proteins come in contact with the nitrocellulose membrane, which binds the protein very strongly on to the surfaces as a band, thus producing a replica of original gels. However, the protein location and detection can only be assessed after immune detection (Figures 6 and 7).

Figure 7: Identification of proteins separated by gel electrophoresis is limited by the small pore size of the gel, as macromolecular probes for protein analysis cannot permeate the gel. The separated proteins are buried in the polyacrylamide gels and therefore further analysis of proteins or their recovery is cumbersome. However, the proteins can be effectively transferred from the gel to the supporting medium by blotting. The presence of SDS facilitates the migration of proteins. Once out of the gel, the proteins come in contact with the nitrocellulose membrane, which binds the protein very strongly on to the surfaces as a band, thus producing a replica of original gels. A variety of analysis involving immuno-chemical analysis can be carried out for the detection of protein bands.
Stage III: Immunodetection
The transferred proteins are bound to the surface of the nitrocellulose membrane and are accessible for reaction with immunochemical reagents.
1. Blocking nonspecific sites: After electrotransfer, the NC membrane was placed in a petridish and the non-specific sites i.e. the unabsorbed sites on NC membranes were blocked by agitating the membrane for 1 h in serum diluent buffer. After incubation, the membrane was washed thrice with rinse buffer [8,9].
2. Labeled antibody (anti-human IgG HRP-conjugate): After washing, the membrane was agitated for 1 h in 10 ml of 1:1000 diluted anti-human IgG HRP conjugate Excess antibody enzyme conjugate was removed by washing the membranes thrice with rinse buffer.
3. Substrate solution: After washing, the membrane was agitated in substrate solution (TMB/H2O2) till blue/grey colored bands were observed. The blot was photographed for permanent record.
Chemicals and reagents
Antigen: Fraction no 2 (serum samples1, 2 and 3)
Phosphate buffer saline (PBS) pH 7.4 (equilibration buffer):(20X/1000 ml):
a. Sodium chloride=180 g
b. Potassium dihydrogen orthophosphate=2.88 g
c. Disodium hydrogen orthophosphate 2H2O=17.78 g
d. 1000 ml of distilled water
180 g sodium chloride, 2.88 g of dipotassium hydrogen orthophosphate and 17.78 g of disodium hydrogen ortho phosphate was dissolved in about 800 ml of distilled water. After adjusting the pH to 7.4 with 10 N sodium hydroxide, the volume was made up to 1000 ml with distilled water. At the time of use the buffer was diluted 20 times (e.g. 50 ml of 20 X PBS+950 ml of distilled water).
Rinse buffer: 50 ml of 20X PBS was diluted to 1000 ml with distilled water. After mixing, 1 ml of tween 20 was added to it.
Serum diluent buffer: 1 g of egg albumin was dissolved in 100 ml of rinse buffer.
Labeled antibody: Goat anti-human fluorescein isothiocyanate (FITC) was purchased from Bangalore Genie Pvt Ltd Bangalore India.
Other requirements:
1. Nitrocellulose membrane strips
2. Micro pipettes 2-20 and 200-1000 μl and tips.
3. Glass pipettes 5 ml and 10 ml.
4. Glass beakers and conical flasks
5. Measuring cylinders
6. Wash bottle
7. Petridish
8. Shaker
9. UV cabinet
Method
2-5 μl of the antigen (Fraction no.2, serum samples 1, 2 and 3) was spotted on nitrocellulose membrane strips. After air drying, the unadsorbed (non-specific) sites on the membrane was blocked by agitating the membrane in 10ml blocking buffer (taken in a petridish) for 1 h on a shaker. After incubation, excess blocking buffer was removed by washing the membrane 3 times with rinse buffer (3×5 min each). After washing with rinse buffer, the membrane was agitated in 1:200 diluted goat anti-human IgG-FITC for 30 min on a shaker. Excess goat anti-human IgG-FITC conjugate was removed by washing the membranes thrice with rinse buffer (3×5 min each). After air drying, the membrane was placed in a UV cabinet to observe the fluorescence spot developed [10-12].
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE)
Chemicals and Reagents:
2% Agarose in 0.9% Saline:
a. 2 g of agarose
b. 0.9 g of sodium chloride
c. 100 ml distilled water
2 g of agarose and 0.9 g of sodium chloride were added to 100 ml of distilled water and the contents were boiled till agarose dissolved completely and the solution was clear.
6N HCl:
a. 48.7 ml distilled water
b. 51.3 ml of conc. HCl
To 48.7 ml distilled water, 51.3 ml of conc. HCl was added.
1.5M Tris-HCI pH 8.8:
a. 18.171 g of Tris base
b. 100 ml distilled water
18.171 g of Tris base was dissolved in about 70 ml of distilled water. After adjusting the pH to
8.8 with 6N HCl, the volume was made up to 100 ml with distilled water(*Initial pH of Tris-base was 10.68; a total of 4.6 ml of 6N HCl was required to get down the pH from 10.68 to 8.8).
0.5M Tris-HCl pH 6.8:
a. 6.057 g of Tris base
b. 100 ml distilled water
6.057 g of Tris base was dissolved in about 70 ml of distilled water. After adjusting the pH to 6.8 with 6N HCl, the volume was made up to 100 ml with distilled water (*Initial pH of Tris-base was 10.47; a total of 9.1 ml of 6N HCl was required to get down the pH from 10.47 to 6.8).
Acrylamide: N, N'-methylene bis acrylamide (normally referred to as bisacrylamide)
a. 30 g of acrylamide
b. 0.8 g of bis- acrylamide
c. 100 ml distilled water
30 g of acrylamide and 0.8 g of bis-acrylamide was dissolved in about 60 ml of distilled water and then the volume was made up to 100 ml with distilled water. The solution was filtered through a Whatman filter paper and then stored in brown bottle.
Note: Acrylamide and Bis-acrylamide are slowly converted during storage to acrylic acid, Bis -acrylic acid. This deamination reaction is catalyzed by light and alkali. Check the pH of the solution, it should be 7.0 or less, and store the solution in dark bottles. Fresh solution should be prepared for every few months. Pre packed, pre mixed stock solutions are commercially available.
10% Sodium dodecyl sulfate (SDS):
a. 10 g of SDS
b. 100 ml distilled water
10 g of SDS was dissolved in about 80 ml of distilled water and then the volume was made up to 100 ml with distilled water. Note: SDS is an irritant, while weighing avoid contact with skin or inhalation.
10% ammonium per sulfate (APS):
a. 1 g of Ammonium per sulfate
b. 10 ml distilled water
1 g of Ammonium per sulfate was dissolved in about 8 ml of distilled water and the volume was made up to 10 ml with distilled water. Aliquots of 0.5 ml were pipetted out in eppendorf tube. The vials were frozen at -20°C. Just before use, one vial was thawed and used for experiment.
Note: APS has to be prepared freshly. An alternative method is to prepare and store frozen.
N, N, N', N'-tetramethylethylene di amine (TEMED): Commercially available.
0.05% Bromophenol Blue
a. 50 mg of bromophenol Blue
b. 100 ml distilled water
50 mg of bromophenol Blue was dissolved in 100 ml of distilled water.
β-Mercaptoethanol: Commercially available.
Glycerol: Available commercially. Note: As an alternative to glycerol 40% sucrose can be used.
Sample buffer:
a. 0.5M Tris -HCI pH 6.8=1 ml
b. Glycerol=1 ml
c. 10% SDS=l ml
d. β-Mercaptoethanol=0.2 ml
e. 0.05% Bromo phenol Blue=0.2 ml
Ingredients a-e were mixed together and then stored in a screw cap vial.
Leveler:
To 1 ml of distilled water 10 μl of 10% SDS was added.
Electrode buffer (reservoir buffer): Tris-Glycine pH 8.3
a. 6 g of Tris-base
b. 28.4 g glycine
c. 10 ml 10% SDS
d. 1000 ml distilled water
6 g of Tris-base was dissolved in about 700 ml of distilled water. The pH of the solution was adjusted to 8.3 with gradual addition of solid glycine. After adjusting the pH, 10 ml of 10% SDS was added and the volume made up to 1000 ml with distilled water. (*28.4 g of glycine was required to adjust the pH to 8.3).
Note: Buffer used in the apparatus with small volume buffer components should be discarded after each run because pH changes occur as a result of the electrolysis of water during electrophoresis. When compartment volumes are >700 ml, switching, polarity of the electrophoretic cell after each run allows use of the same buffer for up to 4 runs.
Staining solution: 0.25% Coomassie Brilliant Blue R250 (in methanol: Acetic acid: water: 50:10:40)
a. 50 ml methanol
b. 10 ml acetic acid
c. 40 ml distilled water
d. 250 mg Coomassie Brilliant Blue R 250
Methanol, acetic acid and water were mixed in the ratio of 50: 10: 40 and then 250 mg of Coomassie Brilliant Blue R250 was dissolved in it. The stain solution was filtered through a plug of cotton. Note: The stain solution can be used 5-6 times, if stored properly.
Destaining solution:
a. 50 ml methanol
b. 10 ml acetic acid
c. 40 ml distilled water
Methanol: acetic acid: water was mixed in the ratio 50: 10: 40 and the solution were stored at room temperature in a screw cap bottle.
Note: The de-stained solution can be re used once or twice if the solution is passed through activated charcoal powder. (Coomassie Brilliant Blue R250 adsorbs onto the charcoal and the methanol: acetic acid: water becomes clear of the stain).
Fixative: 5% Trichloro acetic acid:
a. 5 g of Trichloro acetic acid
b. 100 ml of distilled water
5 g of Trichloro acetic acid was dissolved in 100 ml of distilled water and the solution was stored at room temperature. Note: This solution can be used only once. Before staining the gels are immersed in the fixative to guard against diffusion of separated components.
Silver staining: (all reagents are to be prepared fresh and in double distilled water)
a. Fixative: Methanol: acetic acid: water:: 50: 12: 38 containing 75 μl of formaldehyde
Methanol, acetic acid and water were mixed in the ratio 50:12:38. To this 75 μl of formaldehyde was added.
b. 50% Ethanol: Ethanol and distilled water were mixed in 1:1 ratio (75 ml+75 ml).
c. 0.02% Sodium thiosulfate: 20 mg of sodium thiosulfate was dissolved in about 80 ml of distilled water and then the volume was made up to 100 ml with distilled water.
d. 0.2% Silver nitrate, containing 0.75% formaldehyde: 200 mg of silver nitrate was dissolved in about 80 ml of distilled water and then the volume was made up to 100 ml with distilled water. To this 75 μl of formaldehyde was added.
e. Developer: 6% sodium carbonate containing 0.05% formaldehyde and 4 ml of 0.02% sodium thiosulfate
6 g of sodium carbonate was dissolved in about 70 ml of distilled water. To this 4 ml of 0.02% sodium thiosulfate was added along with 50 μl of formaldehyde and the volume was made up to 100 ml with distilled water and the solution was stored in dark at room temperature.
f. Stop solution 5% Citric acid: 5 g of Citric acid was dissolved in about 80 ml distilled water and then the volume was made up to l00 ml with distilled water.
Samples
a. Pure human IgG
b. Fraction no 2 ( serum samples 1, 2 and 3)
Other requirements
1. Glass beakers 50, l00, 250, 500 and 1000 ml
2. 5 ml, 10 ml glass pipettes
3. 2-20, 20-200 and 200-1000 μl micro pipettes and tips
4. Tissue paper, blotting paper, Whatman No.1 filter paper
5. Dissection needle
6. Hot plate
7. Magnetic stirrer and magnetic bar
8. pH meter
9. Eppendorf tubes (1.5 ml)
10. Gel staining trays & Float
11. Semi log graph sheets, pencil, eraser, scale
12. Adhesive tapes
13. SDS-PAGE electrophoresis unit
14. Power pack
Method
Samples to be analyzed on SDS-PAGE were mixed with sample buffer in a 1:1 ratio. After mixing, the samples were boiled for 10 min. After selecting the appropriate thickness spacers and comb, the glass sandwich was assembled as follows: After placing the glass plate on a leveled surface, silicone grease was applied to the spacers and the spacers were placed on the left and right edge of the plate. After the spacers were fixed on the glass plate the notched plate was placed on the rectangular plate. The glass plate assembly was then clamped in order to keep the assembly tightly together in position. After clamping, the lower end of the glass plate was sealed with 2% agarose in 0.9% saline (Agarose was poured into a boat or gel casting tray and then the assembly was placed in the boat). The whole setup was left undisturbed for the gel to set/solidify. Casting of separating gel and stacking gel was carried out in the gel casting unit (Figure 8).

Figure 8: Small vertical gel Electrophoresis system consists of electrophoresis unit.
Preparation of Separating Gel (12%)
After the agarose gel has set, 5-10 ml of the separating gel was prepared by mixing the ingredients given Table 1.
| Relative binding affinity IgG | ||
|---|---|---|
| Species | Polyclonal | |
| Rabbit | ++++ +++ | |
| Human IgG | ++++ ++++ | |
| Dog | +++ | |
| IgM | —- + | |
| IgD | —- + | |
| IgA | —- + | |
| Cow | ++ ++++ | |
| Horse | ++ ++++ | |
| Goat | - ++ | |
| Guinea pig | ++++ ++ | |
| Sheep | +/- ++ | |
| Pig | +++ +++ | |
| Rat | +/- ++ | |
| Mouse | ++ ++ | |
| Chicken | —- + | |
| Species/ Subclass Protein-A Protein-G | ||
| Monoclonal | ||
| Human | Mouse | Rat |
| IgG 1 ++++ ++++ | IgG 1 + ++++ | IgG 1 —- + |
| IgG 2 ++++ ++++ | IgG 2a ++++ ++++ | IgG 2a —- ++++ |
| IgG 3 —- ++++ | IgG 2b +++ +++ | IgG 2b —- ++ |
| IgG 4 ++++ ++++ | IgG 3 ++ +++ | IgG 2c + ++ |
—- (weak or no binding) ++++ (Strong binding)
Table 1: Relative Affinity of Immobilized Protein-A and Protein-G for Various Antibody Species and Subclasses of Polyclonal and Monoclonal IgG.
After mixing the solution, it was poured into the chamber between the glass plate sandwich assemblies. Immediately after pouring the separating gel mix, 400 μl of the leveler solution was added on the top of the separating gel to even the surface. The setup was left undisturbed for the gel to polymerize. After the separating gel polymerized, the leveler solution was removed by blotting with filter paper.
Preparation of Stacking Gel
After blotting out the leveler 2.5 ml of the stacking gel was prepared by mixing the ingredients given in Table 2. And then it was poured over the separating gel. Immediately after pouring, glycerol applied comb was inserted into the stacking gel. (Glycerol prevents sticking of the gel to the comb), for the formation of wells into which samples are to be loaded.
| Fraction No. | Protein Content | ||
|---|---|---|---|
| Serum sample-1 (mg/ml) | Serum sample-2 (mg/ml) | Serum sample-3 (mg/ml) | |
| 1 | 0.49 | 0.11 | 0.23 |
| 2 | 2.00 | 1.05 | 1.28 |
| 3 | 0.98 | 0.31 | 0.78 |
| 4 | 0.70 | 0.15 | 0.59 |
Table 2: Protein Content in Protein-A Fraction.
Once the stacking gel polymerized the glass plate assembly with the separating and stacking gels was clamped to the electrophoresis apparatus (Care was taken while clamping the plates i.e. the notched plate was facing towards the inner side and the rectangular facing outside). After clamping the plates, electrode buffer was filled into both the upper and lower buffer reservoirs. After the cathodic and anodic buffers were filled, the electrophoresis apparatus was connected to the power pack via connecting cords (Note: black was connected to cathode and red to anode, since proteins are negatively charged they migrate towards anode).
After connecting the cords, the protein samples (according to the method of staining (Coomassie Brilliant Blue R250: 10-25 μl/well, or Silver staining: 5-15 μl/well) were loaded into the wells of stacking gel. After loading the samples, the power pack was switched on and the power set to 70 V. After the tracking dye entered the separating gel, the voltage was increased to 100 V and electrophoresis was carried out till the tracking dye reached the other end of the gel. After the completion of the run, the gel was carefully removed from in between the glass plates and subjected to either Coomassie Brilliant Blue R 250 or silver staining.
Staining the gels
Coomassie Brilliant Blue R250 staining: After the completion of the run, the gel was carefully removed from in between the glass plates and it was fixed in 5% TCA for 1 h. (TCA guards against diffusion of separated protein components). After fixing for 1h, the gel was stained in 0.25% Coomassie Brilliant Blue R 250 solution for 3 h. After staining, the gel was destained overnight to remove the background stain. The gel was then photographed for permanent record.
Silver staining: The proteins separated on gels can be visualized by staining either with Coomassie Brilliant Blue R-250 or Amido black l0 B dye. The drawback of the above dyes is that it detects a band containing 1 μg of the protein. In many occasions, the available protein for electrophoresis is so small or some proteins occur in minute amounts that the detection becomes extremely difficult with these dyes. Under such circumstances a higher sensitive detection system is required. Silver staining is a very useful method in this regard with about 100 fold greater sensitivity over dye staining. It is comparable in sensitivity to autoradiography of labeled polypeptides. There are different methods described by different workers for silver staining. The method given below is very simple and rapid.
Principle: The amino acids particularly aromatic in the protein reduce silver nitrate and form complexes with metallic silver of yellowish-brown to brown color.
Method: After electrophoresis, the gel was fixed in methanol: acetic acid: water (50: 12:38) containing 75 μl of 37% formaldehyde for 1 h or overnight with uniform shaking. Once the gel is removed from the fixer further steps are to be carried out without any delay. After fixing, the gel was treated with 50% ethanol 3 times (3xl0 min each). After ethanol treatment, the gel was impregnated with 0.02% sodium thiosulfate exactly for 1 min (This step is very crucial as the band and background staining depends on this step). After sodium thiosulfate treatment, the gel was washed 3 times with distilled water (3×l min). After washing, the gel was agitated in 0.2% silver nitrate containing 0.075% formaldehyde for 20 min on a shaker. After silver nitrate treatment, the gel was washed 3 times with distilled water (3×1 min each). After washing, the gel was agitated in the developer till the bands developed (Care was taken such that the gel did not takes up the background stain). Immediately after the appearance of the bands the gel was washed several times with excess of distilled water or alternatively the gel was placed in the stop solution (5% citric acid). After staining, the gel was photographed for permanent record [13-17].
Native–PAGE (Activity staining of the enzyme HRP): Native- PAGE analysis of HRP was essentially carried out as described for SDSPAGE methodology section but with slight modification. Separating and stacking gel was prepared by mixing the ingredients given Tables 3 and 4.
| Ingredients | 7.5% | 10% | 12% | 15% | |
|---|---|---|---|---|---|
| 1 | 1.5M Tris HCl pH 8.8 (ml) | 2.5 | 2.5 | 2.5 | 2.5 |
| 2 | Acrylamide: Bisacrylamide | 2.5 | 3.33 | 4.0 | 5.0 |
| 3 | Distilled water | 4.83 | 4.0 | 3.33 | 2.33 |
| 4 | 10% SDS | 0.1 | 0.1 | 0.1 | 0.1 |
| 5 | TEMED | 10 | 10 | 10 | 10 |
| 6 | 10% APS | 75 | 75 | 75 | 75 |
Table 3: Preparation of Separating Gel (10 ml).
| Ingredients | ||
| 1 | 0.5M Tris Hel pH 6.8 | 0.75 ml |
| 2 | Acrylamide-Bisacrylamide | 1.0 ml |
| 3 | Distilled water | 3.15 ml |
| 4 | 10% SDS | 50 μl |
| 5 | 10% APS | 45 μl |
| 6 | TEMED | 10 μl |
Table 4: Preparation of Stacking Gel (5 ml).
The sample (HRP) was diluted with equal volume of sample buffer and then 30-40 μg of the sample was loaded in to the well. Electrophoresis was carried out at 100 V. After completion of electrophoresis, the gel was stained with substrate solution (DAB system) till the development of colored band.
Conjugation of horseradish peroxidase to antibodies: The present study that describes the conjugation of horseradish peroxidase to anti-human IgG was initiated because; direct conjugation of enzymes to antibodies has greatly simplified the development and performance of many different types of immunoassays. Horseradish peroxidase (HRPO) conjugates are useful in all types of immunological assays e.g., Horseradish peroxidase–antibody conjugates can be used in ELISA (enzyme-linked immunosorbent assay) and western blotting [18].
Result: Conjugation of horseradish peroxidase (HRPO) to antibody (IgG) was essentially carried out by periodate oxidation method.
The method involves three chemical steps:
1. Sodium periodate (NaIO4)-oxidation of the carbohydrate side chains of HRPO
2. Schiff base formation between activated peroxidase and amino groups of the antibody and
3. Sodium borohydride (NaBH4) reduction of the Schiff base to form a stable conjugate.
After preparing the antibody (IgG) horseradish peroxidase conjugate, as a preliminary study, the efficacy of the enzyme-labeled antibody was tested by direct dot-ELISA. As shown in Figure 9, the titer of the anti-human IgG–HRP conjugate was found out to be 1:4000.

Figure 9: The efficacy of the enzyme-labeled antibody was tested by direct Dot-ELISA. Human IgG were coated on to four nitrocellulose strips that were labeled 1:1000, 1:2000, 1:4000 and 1:8000. After air drying, the unadsorbed sites on the membrane were blocked with blocking buffer. After blocking, the membranes were agitated in different dilutions of the HRP conjugate (1:1000, 1:2000, 1:4000 and 1:8000). Excess HRP conjugate was removed by washing the membrane. The membrane was then agitated in the substrate solution, tetra methyl benzidine–H2O2 (TMB/H2O2) till blue/grey colored spot developed.
The HRP conjugate was used for the following experiments:
• ELISA
• Western blot analysis
To carry out ELISA and western blot experiments a suitable antigen is required. Towards this goal, IgG from three different human serum samples were purified on protein-A affinity chromatography.
Purification of human IgG employing protein-A affinity chromatography
A protein must be purified before its structure and the mechanism of its action can be studied. However, because proteins vary in size, charge, and water solubility, no single method can be used to isolate all proteins. To isolate one particular protein from the estimated 10,000 different proteins in a cell is a daunting task that requires methods both for separating proteins and for detecting the presence of specific proteins. Any molecule, whether protein, carbohydrate, or nucleic acid, can be separated, or resolved, from other molecules on the basis of their differences in one or more physical or chemical characteristics. The larger and more numerous differences between two proteins, the separation of the proteins is easier and more efficient. The two most widely used characteristics for separating proteins are size, defined as either length or mass, and binding affinity for specific ligand.
Affinity chromatography
Affinity chromatography is the process of bio-selective adsorption and subsequent recovery of a compound from an immobilized ligand. The ability of proteins to bind specifically to other molecules is the basis of affinity chromatography. In this technique, ligand molecules that bind to the protein of interest are covalently attached to the beads used to form the column. Ligand can be enzyme substrates or other small molecules that bind to specific proteins. In a widely used form of this technique, antibody-affinity chromatography, the attached ligand is an antibody specific for the desired protein. An affinity column will retain only those proteins that bind the ligand attached to the beads; the remaining proteins, regardless of their charges or masses, will pass through the column without binding to it. The proteins bound to the affinity column are then eluted by adding an excess of ligand or by changing the salt concentration or pH. The ability of this technique to separate particular proteins depends on the selection of appropriate ligand. The affinity chromatography is the most acceptable method for purification of biomolecules. Theoretically affinity chromatography should give a fairly pure sample in a single step.
Protein-A has an affinity to bind to IgG of various species. This property of protein-A was employed for the purification of IgG from serum of different human serum samples in our studies.
Staphylococcal protein-A
Staphylococcal protein A is an IgG binding protein found in the bacterial cell wall of Staphylococcus aureus. Staphylococcal protein-A binds to most mammalian IgG and can be used for detecting or purifying such antibodies. Affinity chromatography on staphylococcal protein A column is extensively used for purifying monoclonal and polyclonal antibodies.
Protein A: Staphylococcal protein A is a 42 Kilo Dalton protein, exists both in secreted and membrane-associated form. There are 4 Fc binding, highly homologous regions each constituting of 58 to 62 amino acids. These regions are consecutively arranged from the N terminal part of protein. The residual C terminal part is approximately 150 residues long differs to a great extent with respect to primary and secondary structures from the 4 active regions. Furthermore, it is suggested that the protein is bound to the bacterial cell-wall structure through this C terminal part. The binding of protein-A has been well documented for immunoglobulin’s from a variety of mammalian species and for immunoglobulin’s IgM and IgA as well.
Advantages of protein-A column:
• High immunoglobulin binding capacity
• High flow rate with good resolution
• Highly stability to all physical changes
• The dissociation constant Kd=10-4 to 10-8 permits recovery under mild conditions hence called high performance affinity chromatography (HPAC)
Results: IgG from three different human serum samples were purified on protein-A column. The absorbance values for eluted fractions are given in Table 5. Serum sample (0.5 ml serum+0.5 ml equilibration buffer) was loaded on protein A column that was equilibrated with equilibration buffer (PBS pH 7.4). 1ml fractions were collected and A280 for each fraction was read in a UV–Vis spectrophotometer and absorbance values are given in the Table 6 and the elution profiles are shown in Figure 10. As evident from Figure 11, IgG from human serum samples 1, 2 and 3 fractionated into single peak. Among the different fractions in the peak, fraction no.2 showed the highest absorbance value in all the 3 samples. Since peak fraction no.2 showed highest absorbance values, further experiments were carried out with this fraction only.
| Formulation For Preparation Of Different Percentages Of Separating Gel (10ml) | |||||
|---|---|---|---|---|---|
| Ingredients | 7.5 % | 10 % | 12 % | 15 % | |
| 1 | 1.5M Tris HCl pH 8.8 (ml) | 2.5 ml | 2.5 ml | 2.5 ml | 2.5 ml |
| 2 | Acrylamide: Bisacrylamide | 2.5 ml | 3.33 ml | 4.0 ml | 5.0 ml |
| 3 | Distilled water | 4.93 ml | 4.10 ml | 3.43 ml | 2.43 ml |
| 4 | TEMED | 10 μl | 10 μl | 10 μl | 10 μl |
| 5 | 10% APS | 75 μl | 75 μl | 75 μl | 75 μl |
Table 5: Preparation of Separating Gel and Stacking for Native-Page Analysis.
| Fraction. No | Serum sample-1 A280 | Serum sample-2 A280 | Serum sample-3 A280 |
|---|---|---|---|
| 1. | 0.69 | 0.16 | 0.32 |
| 2. | 2.81 | 1.47 | 1.80 |
| 3. | 1.38 | 0.44 | 1.10 |
| 4. | 0.98 | 0.21 | 0.83 |
| 5. | 0.71 | 0.15 | 0.50 |
| 6. | 0.54 | 0.13 | 0.43 |
| 7. | 0.40 | 0.12 | 0.31 |
| 8. | 0.28 | 0.10 | 0.19 |
| 9. | 0.17 | 0.07 | 0.11 |
| 10. | 0.07 | 0.03 | 0.06 |
Table 6: Absorbance Values for Protein-A Eluted Fractions.

Figure 10: Elution Profile for IgG Isolated from Different Human Serum Samples.

Figure 11: Variations in the Enzyme-Linked Immunosorbent Assay (ELISA) Technique.
In order to confirm the presence of IgG in fraction no.2, ELISA and western blot experiments were performed. These experiments were carried out with anti-human IgG HRP conjugate that was prepared in our laboratory.
Enzyme Linked Immunosorbent Assay (ELISA): Modern medicine is dependent up on various tools of investigation for arriving at a correct diagnosis. Most of the in-vitro methods rely up on detection and accurate measurement of a particular component in different physiological fluids and tissues of the human body. Immunoassay exploits the antigen-antibody reaction to achieve this end.
Antigen: An “antigen” (Ag) is defined as any foreign substance, which is capable of eliciting an immune response (humoral, cellular, or most commonly, both) in the host. Lately the term “immunogen” is used more frequently instead of antigen. The most potent immunogens are macromolecular proteins, but polysaccharides, nucleic acids, lipids, synthetic polypeptides and other synthetic polymers such as polyvinyl pyrolidone can act as antigens. The antigenicity of a molecule depends on a number of properties listed below:
1. Foreignness.
2. Molecular size.
3. Chemical complexity.
4. Genetic constitution of the animal.
5. Method and route of antigen administration.
6. Antigen concentration.
7. Amino acid composition.
Antibody (Immunoglobulin’s): Antibodies are heterogeneous group of globulins mainly found in the serum. Antibody is a glycoprotein that is produced in response to introduction of an antigen and which has the ability to combine with the antigen that stimulated its production.
Since the antibodies are glycoproteins, they can also act as antigens when inoculated in to a different mammalian species. There are five types of immunoglobulin’s (Igs). The classification of the immunoglobulin’s is based on the type of “heavy chain” present in the antibody. Thus, immunoglobulin’s containing heavy chains γ, μ, α, δ and ε are called immunoglobulin-G (IgG), immunoglobulin-M (IgM), immunoglobulin-A (IgA), immunoglobulin-D (IgD), and immunoglobulin-E (IgE) respectively. In addition, several sub-classes are also known. When an individual is immunized against single antigen, specific antibodies are produced which usually consist of a mixture of IgM, IgG and IgA. The amount of each immunoglobulin’s produced depends on the nature of the antigen and the stage of immunity. Specificity and sensitivity are the two major features of antigen-antibody reactions. Of the various immunoassays was the turning point in the accurate measurement of substances at ultra-lowlevel. The radioimmunoassay suffers from certain inherent drawbacks such as:
1. Radioimmunoassay requires constant supply of radioisotopes.
2. Short half-life of radiolabel.
3. Requirement of highly skilled technicians.
4. Expensive infrastructure (i.e. Scintillation counters/ spectrometers are required.
5. Potential health hazards associated with routine use of radioactive material (i.e. facility for radioactive substances have also to be protected against radiation hazards).
6. Problem of disposal of radioactive waste.
These drawbacks of radioimmunoassay have been circumvented by development of an similarly sensitive and versatile technique in which in place of radioactive compound, enzymes have been used as ‘labels’ or ‘markers’ in immunoassays. Immunoassays in which either the primary or the secondary antibody is coupled to an enzyme are referred to as enzyme immunoassays. Enzyme immunoassays while incorporating almost all the benefits of radioimmunoassay, Assays, provide additional advantages in terms of ease of operation, safety of personnel and cost effectiveness.
The term “enzyme linked immunosorbent assay” (ELISA) was coined in 1971 by Engvall and Peralman for this enzyme based technique. Over the last two decades, application of enzyme immunoassays has expanded quite dramatically. Enzyme immunoassays are today playing an increasingly important role in diagnostics and research laboratories the world over. The developments in automation of immunoassays and monoclonal antibody technology have enabled introduction of a variety variations in the basic ELISA procedures to fulfill the requirements of different test systems. The application of monoclonal antibodies in ELISA protocols has enhanced specificity of the test. ELISA is presently one of the most commonly used tests for the detection of antigens and antibodies. In addition ELISA can also be employed for the quantitation of antigens and antibodies. There are a variety of modifications of this test and these are collectively known as enzyme immunoassays (EIA).
The basic feature of this group of assays is as follows: One of the immunereagent (usually antigen at times antibody) is immobilized through adsorption on the solid phase support (usually polyvinyl chloride or polystyrene) in such a way that there is no loss to its activity. The second immunoreagent (either primary or secondary antibody) is linked to an enzyme in a way that there is no loss either to immuno reactivity or to the enzyme activity.
All assays are carried out in 96 well flat bottomed polystyrene or polyvinyl chloride plates.
1. Antigen coating: 100-200 μl of the antigen (usually antigen at times antibody) at a concentration of 5-10 μg per ml in 0.1 M sodium carbonate–bicarbonate buffer, pH 9.6 is added into wells of the flat bottomed microtiter plate. After incubation for appropriate time period at 37°C or room temperature or at 4°C, excess antigen or antibody is removed by washing the wells 5 times with rinse buffer.
2. Blocking nonspecific sites: After washing, non-specific sites are blocked by addition of 300 μl serum diluent buffer into each well and the plate is incubated for 1 h at 37°C. After incubation, wells are washed 5 times with rinse buffer to remove excess blocking.
3. Labeled antibody: After washing, 100-200 μl of appropriately diluted antibody enzyme conjugate is added to wells that are coated with antigen. After incubation for appropriate time period at 37°C or room temperature or at 4°C, excess antibody enzyme conjugate is removed by washing the wells 5 times with rinse buffer.
4. Substrate solution: After washing the wells, 100-200 μl of the chromogenic substrate (depending on the enzyme conjugated to the antibody a suitable chromogenic substrate is added to each well) for e.g. if the enzyme Horseradish peroxidase is conjugated to the antibody the chromogenic substrate TMB/H2O2 used or if the enzyme Alkaline phosphatase is conjugated to the antibody then substrate paranitrophenol phosphate is used) and the plate is incubated at room temperature in dark till the development of the colour. After colour development, the reaction is stopped by adding 100 μl of either 2N HCl or 2N H2SO4 into each well and the colour developed is read at 450 nm for TMB/H2O2 or at 405 nm for para nitrophenol phosphate in a micro titter plate auto reader (ELISA reader) or in a colorimeter.
If the two immuno reagents (antigen with its specific antibody) have bound to each other, colour will develop because of the presence of the linked enzyme; If not there will be no colour development due to the lack of the linked enzyme.
Assays for Antibody
Direct method: Salient points of the procedure for direct method are briefed below:
A suitable antigen (antigen in the given sample) is allowed to adsorb on to the wells of micro titer plate. After antigen adsorption, the non-specific sites are blocked by the addition of blocking or serum diluent buffer into the wells. After incubation, the enzyme linked antibodies (Antibody enzyme conjugate) specific for the antigen is added to the wells, and allowed to incubate at 37°C. During this step if the antibody conjugated to the enzyme is specific to the antigen it binds to the adsorbed antigen in the micro titer plate wells. After incubation, a chromogenic enzyme substrate is added to the wells and the plates are incubated till the development of the colour. The reaction is stopped by addition of 2 N HCl and the colour intensity is measured photometrically. (In an ELISA reader).
Indirect method: This method is largely used to measure antibodies in almost all infections (including HIV). The method has become popular because it requires only a single conjugate, for example enzyme labeled anti-human globulins. Different immunoglobulin’s classes can be detected differentially by using class specific conjugates (IgG, IgM, and IgA).
Salient points of the procedures for indirect method are briefed below:
A suitable antigen (antigen in the given sample for e.g. antigen Gp120 in the case of HIV detection) is allowed to adsorb on to the wells of micro titer plate. After antigen adsorption, the non-specific sites are blocked by the addition of blocking buffer into the wells. After incubation, in the subsequent step, appropriately diluted serum or any other test sample (serum from HIV patient e.g. which contains antibodies to Gp120 {primary antibody (Ab1)}) is added to an antigencoated microtiter well and allowed to react with the antigen adsorbed to the well. After incubation, the presence of antibody bound to the antigen is detected by adding an enzyme-conjugated secondary anti-isotype antibody (Ab2). After incubation, enzyme-conjugated secondary anti-isotype antibody (Ab2) is added to the wells and the plates are incubated at 37°C. If the patients sera contains antibody to the specific antigen (e.g. in the case of HIV antibodies to Gp120) the primary antibody specifically binds to the adsorbed antigen in the wells and becomes immobilized. When the enzyme-conjugated secondary anti-isotype antibody (Ab2) is added to these wells it binds to the primary antibody. After incubation, a chromogenic substrate specific for the enzyme coupled to secondary antibody is added to the wells and the plates are incubated in dark till the colour develops. The reaction is stopped after incubation time by the addition of 2 N HCl. The amount of colored reaction product that forms is measured by specialized spectrophotometric plate readers, which can measure the absorbance of all of the wells of a 96-well plate in seconds.
Applications of indirect ELISA:
1. Indirect ELISA is the method of choice to detect the presence of serum antibodies against human immunodeficiency virus (HIV), the causative agent of AIDS. In this assay, recombinant envelope and core proteins of HIV are adsorbed as solid-phase antigens to microtiter wells. Individuals infected with HIV will produce serum antibodies to epitopes on these viral proteins. Generally, serum antibodies to HIV can be detected by indirect ELISA within 6 weeks of infection.
2. The indirect immunosorbent assay currently is being used to test for antibodies to rubella virus (German measles), and to detect certain drugs in serum. For example, antigen-coated latex beads are used in the SUDS HIV-1 test to detect HIV serum antibodies in about 10 minutes.
Sandwich ELISA: Antigen can be detected or measured by a sandwich ELISA. In this technique, the antibody (rather than the antigen) is immobilized on a microtiter well. This method may be the most versatile and sensitive for the detection of antigens in mixtures. No purified antigen is required. However, only multivalent antigens with different or repeating epitopes may be detected in this assay since binding of two antibodies to the antigen is required. This requirement is normally not a limitation for proteins, which are almost always multivalent. This method is also referred to as double antibody method.
Salient points of the procedures for indirect method are briefed below:
In this technique, the antibody (rather than the antigen) is immobilized on a microtiter well. The appropriately diluted unlabeled antibody (Capture antibody) is adsorbed on to the 96 well micro titer plates. After antibody adsorption, the non-specific sites are blocked by the addition of blocking buffer into the wells. After incubation, a sample containing antigen (e.g. culture supernatants from antigens produced by recombinant technology) is added and allowed to react with the immobilized antibody. After incubation, a second enzymelinked antibody specific for the same epitope (if multiple copies of the same epitope is present) or a second enzyme-linked antibody specific for different epitope on the antigen is added and allowed to react with the bound antigen. After incubation, a chromogenic substrate specific for the enzyme coupled to secondary antibody is added to the wells and the plates are incubated in dark till the colour develops. The enzyme reaction is terminated by the addition of 2N HCl after the incubation period. The amount of colored reaction product that forms is measured by specialized spectrophotometric plate readers, which can measure the absorbance of all of the wells of a 96-well plate in seconds.
In this method, concentrations of the antigen or antibody in a given test sample can be determined by carrying out the ELISA in presence of different concentrations of the standard antigen or antibody and then constructing a standard curve with antigen or antibody concentrations versus absorbance. From the standard curve the concentration of antigen or antibody in the given sample can be determined.
Result
Serum samples 1, 2, 3 and fraction no.2 from serum samples 1, 2, 3 were tested for the presence of IgG using a direct ELISA. Results are shown in Figure 12. It was observed that anti-human IgG HRP conjugate reacted with wells that were coated with serum samples 1, 2, 3 fraction no.2 from serum samples1, 2, 3 and pure IgG (Figure 13) indicating the presence of IgG in these samples. Anti-human IgG-HRP conjugate failed to react with wells that were coated with rabbit IgG (Figure 13). These results not only confirm for the presence of IgG but also indicated the specificities of anti-human IgG-HRP conjugate for human IgG.

Figure 12: Enzyme Linked Immunosorbent Assay.

Figure 13: Western Blot Analysis.
Highly specific enzyme and antibody assays can detect individual proteins
The purification of a protein, or any other molecule, requires a specific assay that can detect the molecule of interest in column fractions or gel bands. An assay capitalizes on some highly distinctive characteristic of a protein: the ability to bind a particular ligand, to catalyze a particular reaction, or to be recognized by a specific antibody. An assay must also be simple and fast to minimize errors and the possibility that the protein of interest becomes denatured or degraded while the assay is performed. The goal of any purification scheme is to isolate sufficient amounts of a given protein for study; thus a useful assay must also be sensitive enough that only a small proportion of the available material is consumed. Many common protein assays require just from 10-9 to 10-12 g of material.
Chromogenic and light-emitting enzyme reactions
Many assays are tailored to detect some functional aspect of a protein. For example, enzyme assays are based on the ability to detect the loss of substrate or the formation of product. Some enzyme assays utilize chromogenic substrates, which change color in the course of the reaction. (Some substrates are naturally chromogenic; if they are not, they can be linked to a chromogenic molecule). Because of the specificity of an enzyme for its substrate, only samples that contain the enzyme will change color in the presence of a chromogenic substrate and other required reaction components; the rate of the reaction provides a measure of the quantity of enzyme present. Such chromogenic enzymes can also be fused or chemically linked to an antibody and used to “report” the presence or location of the antigen. Alternatively, luciferase, an enzyme present in fireflies and some bacteria, can be linked to an antibody. In the presence of ATP and luciferin, luciferase catalyzes a light-emitting reaction. In either case, after the antibody binds to the protein of interest, substrates of the linked enzyme are added and the appearance of color or emitted light is monitored. A variation of this technique, particularly useful in detecting specific proteins within living cells, makes use of green fluorescent protein (GFP), a naturally fluorescent protein found in jellyfish.
Western Blotting or immune blotting, a powerful method for detecting a particular protein in a complex mixture combines the superior resolving power of gel electrophoresis, the specificity of antibodies, and the sensitivity of enzyme assays. It is so named in view of the previously used nomenclature for nucleic acid blotting procedure i.e. the transfer of DNA fragments on to nitrocellulose paper or nylon membrane is called southern blotting which has been named after its inventor southern. In a similar way RNA molecules can be transferred to nitrocellulose membrane and the technique is known as northern blotting by analogy to southern blotting.
This multistep procedure is commonly used to separate proteins and then identify a specific protein of interest. Two different antibodies are used in this method, one specific for the desired protein and the other linked to a reporter enzyme. Blotting of protein bands allows them to become more accessible for detection and identification using a variety of methods such as immunological detection, autoradiography, analysis of DNA binding proteins, glycoprotein’s, etc. and also the membranes can be stored for much longer period. A number of supporting matrices such as nitrocellulose, diazobenzyl oxymethyl cellulose sheets, nylon membrane and Hydrogen bond membrane etc., are used for this purpose. The use of membrane as a support for protein enables the ease of manipulation, efficient washing and faster reaction during the immuno detection, as proteins are more accessible for reaction.
Blotting is the transfer of resolved proteins from the gel to the surface of a suitable membrane. The transfer of proteins from gels can be achieved by any one of the methods such as simple capillary action, application of vaccum or electrophoretically. Electro blotting is the most efficient and has become a standard method. In this method, the transfer buffer (0.02 M tris-HCl, 0.15 M glycine and 20% methanol) has low ionic strength, which allows electro transfer of proteins. Methanol in the buffer increases the binding of proteins to nitrocellulose and reduces swelling of the gel during transfer. The protein is transferred to the corresponding position on the membrane as on the gel. A mirror image of the gel is formed. However, the protein location and detection can only be assessed after immunodetection.
Result
Fraction no.2 from serum samples 1, 2, 3 was analyzed by western blotting. Results are shown in Figure 13.
Fraction no 2 from serum samples 1, 2, and 3 were analyzed by western blotting. Western blotting is essentially a combination of three techniques viz., electrophoresis (SDS-PAGE), Electrotransfer (protein blotting) and Immunochemical detection (blot development).
Stage I: Separation of proteins on SDS-PAGE: The protein (Fraction no 2 from serum samples 1, 2, and 3) to be analyzed by western blotting is first subjected to separation on a 10-12% separating gel and 6%stacking gel.
Stage II: After electrophoresis the proteins separated on SDSPAGE were transferred to a more stable medium such as nitrocellulose membrane. During transfer the membrane is kept in close contact with the gel containing the separated proteins. At the end of electro transfer, proteins from the gel migrate to the nitrocellulose membrane. The protein is transferred to the corresponding position on the membrane as on the gel. A mirror image of the gel is formed. However, the protein location and detection can only be assessed after immunodetection.
Stage III: Immunodetection: The transferred proteins are bound to the surface of the nitrocellulose membrane and are accessible for reaction with immunochemical reagents. Location of protein bands are detected by immunodetection using a enzyme-labeled (anti human IgG-HRP conjugate) and its suitable substrate (TMB/H2O2).
It was observed that anti-human IgG-HRP conjugate reacted with both the heavy and light chain in fraction no 2 (serum samples 1, 2 and 3) resulting in the lighting up of two bands. These results clearly suggest that the proteins present in fraction no 2 (serum samples 1, 2 and 3) were indeed IgG.
Fluorescent molecules
• Fluorescein, an organic dye that is the most widely used label for immunofluorescence procedures, absorbs blue light (490 nm) and emits an intense yellow-green fluorescence (517 nm).
• Rhodamine, another organic dye, absorbs in the yellow-green range (515 nm) and emits a deep red fluorescence (546 nm). Because it emits fluorescence at a longer wavelength than fluorescein, it can be used in two-color immunofluorescence assays. An antibody specific to one determinant is labeled with fluorescein, and an antibody recognizing a different antigen is labeled with rhodamine. The location of the fluorescein-tagged antibody will be visible by its yellow-green color, easy to distinguish from the red color emitted where the rhodaminetagged antibody has bound. By conjugating fluorescein to one antibody and rhodamine to another antibody, one can, for example, visualize simultaneously two different cell-membrane antigens on the same cell.
• Phycoerythrinis an efficient absorber of light (~30-fold greater than fluorescein) and a brilliant emitter of red fluorescence, stimulating its wide use as a label for immunofluorescence.
Fluorescent-antibody staining of cell membrane molecules or tissue sections can be direct or indirect.
Direct and indirect immunofluorescence staining of membrane antigen
In direct staining, the specific antibody (the primary antibody) is directly conjugated with fluorescein; in indirect staining, the primary antibody is unlabeled and is detected with an additional fluorochromelabeled reagent. A number of reagents have been developed for indirect staining. The most common is a fluorochrome-labeled secondary antibody raised in one species against antibodies of another species, such as fluorescein-labeled goat anti-mouse immunoglobulin. Indirect immunofluorescence staining has two advantages over direct staining. First, the primary antibody does not need to be conjugated with a fluorochrome. Because the supply of primary antibody is often a limiting factor, indirect methods avoid the loss of antibody that usually occurs during the conjugation reaction. Second, indirect methods increase the sensitivity of staining because multiple molecules of the fluorochrome reagent bind to each primary antibody molecule, increasing the amount of light emitted at the location of each primary antibody molecule. Immunofluorescence has been applied to identify a number of subpopulations of lymphocytes, notably the CD4+ and CD8+ T-cell subpopulations.
Result
The presence of IgG in fraction no 2 (serum samples 1, 2 and 3) was further confirmed by immunofluorescence (dot blot method) by using goat anti-human fluorescein isothiocyanate (FITC). It was observed that goat anti-human IgG-FITC reacted with fraction no. 2, from serum samples 1, 2 and 3 to produce a fluorescence spot.
Antigen (fraction no 2 from serum samples 1, 2 and 3) were spotted on nitrocellulose membrane strip. Unadsorbed (non-specific) sites on the membrane were blocked by agitating the membrane in 10 ml blocking buffer. After washing, the membrane was agitated in 1:200 diluted goat anti human IgG-FITC. Excess goat anti human IgG-FITC conjugate was removed by washing. After air drying, the membrane was placed in a UV cabinet to observe the fluorescence spot developed. Results of all the three experiments Viz. ELISA, western blotting and immunofluorescence clearly suggested that the protein present in fraction no.2 (serum samples 1, 2 and 3) was IgG. After confirming the prescience of IgG, the next goal was to determine the purity and molecular weight of purified IgG by SDS-PAGE.
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE)
An important technique for the separation of proteins is based on the migration of charged proteins in an electric field, a process called electrophoresis. These procedures are not generally used to purify proteins in large amounts, because simpler alternatives are usually available and electrophoretic methods often adversely affect the structure and thus the function of proteins. Electrophoresis is, however, especially useful as an analytical method. Its advantage is that proteins can be visualized as well as separated, permitting a researcher to estimate quickly the number of different proteins in a mixture or the degree of purity of a particular protein preparation. Also, electrophoresis allows determination of crucial properties of a protein such as its isoelectric point and approximate molecular weight. The polyacrylamide gel acts as a molecular sieve, slowing the migration of proteins approximately in proportion to their charge-to-mass ratio. Migration may also be affected by protein shape. In electrophoresis, the force moving the macromolecule is the electrical potential, E. The electrophoretic mobility of the molecule, μ, is the ratio of the velocity of the particle molecule, V, to the electrical potential. Electrophoretic mobility is also equal to the net charge of the molecule, Z, divided by the frictional coefficient, f, which reflects in part a protein’s shape.
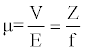
The migration of a protein in a gel during electrophoresis is therefore a function of its size and its shape.
An electrophoretic method commonly employed for estimation of purity and molecular weight makes use of the detergent sodium dodecyl sulfate (SDS).
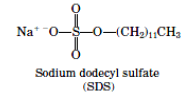
SDS binds to most proteins in amounts roughly proportional to the molecular weight of the protein, about one molecule of SDS for every two amino acid residues. The bound SDS contributes a large net negative charge, rendering the intrinsic charge of the protein insignificant and conferring on each protein a similar charge-to-mass ratio. In addition, the native conformation of a protein is altered when SDS is bound, and most proteins assume a similar shape. Electrophoresis in the presence of SDS therefore separates proteins almost exclusively on the basis of mass (molecular weight), with smaller polypeptides migrating more rapidly. After electrophoresis, the proteins are visualized by adding a dye such as Coomassie blue, which binds to proteins but not to the gel itself. Thus, a researcher can monitor the progress of a protein purification procedure as the number of protein bands visible on the gel decreases after each new fractionation step. When compared with the positions to which proteins of known molecular weight migrate in the gel, the position of an unidentified protein can provide an excellent measure of its molecular weight. If the protein has two or more different subunits, the subunits will generally be separated by the SDS treatment and a separate band will appear for each (Table 7).
| Lane | Molecular Weight marker | Relative mobility (cm) | Rf values | Molecular weight (Daltons) |
|---|---|---|---|---|
| 1 | Fraction no.2 Band-1 Band-2 |
1.2 | 0.38 | 50,000 |
| 1.8 | 0.56 | 24,000 | ||
| 2 | BSA | 1.0 | 0.31 | 66’000 |
| 3 | Lysozyme | 2.2 | 0.68 | 14,000 |
| 4 | Phosphorylase-b Band-1 | 0.7 | 0.22 | 97,400 |
| BSA Band-2 | 1.0 | 0.31 | 66,000 | |
| Ovalbumin Band-3 | 1.6 | 0.50 | 43,000 | |
| Carbonic anhydrase Band-4 | 2.0 | 0.63 | 29,000 | |
| β lactoglobulin Band-5 | 2.1 | 0.65 | 19,400 | |
| Lysozyme Band-6 | 2.2 | 0.69 | 14,300 | |
| 5 | Fraction No.2 Band-1 | 1.2 | 0.38 | 50,000 |
| Fraction No.2 Band-2 | 1.8 | 0.56 | 24,000 |
Table 7: Molecular Weight Determination by SDS-Page.
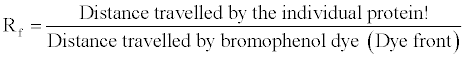
The pioneering work on electrophoresis by A. Tiselius and co-workers was performed in free solution. However, it was soon realized that many of the problems associated with this approach, particularly the adverse effects of diffusion and convection-currents, could be minimized by stabilizing the medium. This was achieved by carrying out electrophoresis on a porous mechanical support, which was wetted in electrophoresis buffer and in which electrophoresis of buffer ions and samples could occur. The support medium cuts down convection currents and diffusion so that the separated components remain as sharp zone. The earliest supports used were filter paper or cellulose acetate strip, wetted in electrophoresis buffer. Nowadays these media are infrequently used; although cellulose acetate still has its use in clinical laboratories for the separation of serum proteins. In particular, for many years’ small molecules such as amino acids, peptides and carbohydrates were routinely separated and analyzed by electrophoresis on supports such as paper or thin-layer plates of cellulose, silica or alumina. Although occasionally still used, such molecules are nowadays more likely to be analyzed by more modern and sensitive techniques. While paper and or thin-layer supports are fine for resolving small molecules, the separation of macromolecules such as proteins and nucleic acids on such supports is poor. However, the introduction of the use of gels as support medium led to a rapid improvement in methods for analyzing macromolecules. The earliest gel system to be used was the starch gel and although this still has some uses, the vast majority of electrophoretic techniques used nowadays involve either polyacrylamide gels or agarose gels. In the case of paper and cellulose acetate electrophoresis, charge on the molecule is the major determinant for its electrophoretic mobility and ultimate separation. The gels in common use, polyacrylamide and agarose, have pores of molecular dimensions whose sizes can be specified. The molecular separations are therefore based on gel filtration as well as the electrophoretic mobilities of the molecules being separated, which shift the equilibrium towards formation of zwitter ions. As zwitter ions do not possess a net charge, they are immobile and carry the current in this region and migrate rapidly in this strong local electric field the pH of the buffer used in the sample is the same buffer that is used in the stacking gel. The Polymerization of Acrylamide and N, N'-Methylene Bis Acrylamide to Form a Cross-Linked Polyacrylamide Gel. Polymerization of acrylamide and N,N'-methylene bisacrylamide to form a cross-linked polyacrylamide gel. The polymerization is induced by free radicals resulting from the chemical decomposition of ammonium persulphate (S2O8-2 2SO4-) or photodecomposition of riboflavin in the presence of traces of O2. In either case TEMED, a free radical stabilizer is usually added to the gel mixture. The physical properties of gel and its pore size are controlled by the proportion of polyacrylamide in the gel and its degree of cross-linking. The most commonly used polyacrylamide concentrations are in the range 3 to 15%. Two different porosity gels commonly used in SDS-PAGE are the stacking gel (high porosity gel) and separating or resolving gel (low porosity gel).
When the current is switched on, all the ionic species have to migrate at the same speed otherwise there would be a break in the electrical circuit. Glycine in the upper buffer reservoir exists in two forms; as zwitterions, which does not have a net charge, and as a glycinate anion with a charge of minus one.
When the power is switched on, chloride, protein and glycinate ions begin to migrate towards the anode. Upon entering the stacking gel, the glycinate ion encounters a condition of low pH, which shifts the equilibrium towards formation of zwitter ions. As zwitter ions do not possess a net charge, they are immobile. This immobility of glycine zwitter ions to migrate in to the stacking gel coupled with high mobility of the chloride ions creates a very high localized voltage gradient between the leading chloride and the trailing glycinate ions. Since proteins have their mobility intermediate between the trailing and the leading ions, they carry the current in this region and migrate rapidly in this strong local electric field. The proteins, however, cannot overtake the chloride ions, as the strong local field exists only between the chloride and the glycinate ions. As a result the proteins migrate quickly until they reach the region rich in chloride ions and then drastically slow down. The result is that the three species of interest adjust their concentrations so that [Cl]>[protein-SDS]>[glycinate]. There is only a small quantity of protein-SDS complexes, so they concentrate in a very tight, sharp band between glycinate and Cl- (Chloride ions) (Chlorideions) boundaries. (i.e., the faster migration of proteins results in piling or stacking up of the protein samples into a tight sharp disc). It is in this form that the macromolecules enter the running gel. The smaller pores of the separating gel retards the movement of the sharp band of the macromolecules for a long enough time for the glycinate anions to catch up. (The larger pores in the stacking gel do not have any sieving effect therefore the macromolecules migrate faster without any hindrance however, when they reach the separating gel the pores are numerous and of a smaller diameter imparting molecular sieving property to the gel therefore the macromolecules cannot migrate with the same speed as they did in the stacking gel as a result the proteins pile or stack up into a tight sharp disc).
Once the glycinate ions reach the separating gel it becomes more fully ionized in the higher pH environment its mobility increases. (The pH of the stacking gel is 6.8 that of the separating gel is 8.8). Thus the interface between glycinate and Cl- (Chloride ions) leaves behind the protein-SDS complexes which are left to electrophoresis at their own rates. (Upon entering the separating gel the glycinate ions encounter a condition of high pH [pH of the separating gel buffer is about 2 pH units higher than that of the stacking gel] which shifts the equilibrium towards formation of glycinate anion). The negatively charged protein- SDS complexes now continue to move towards the anode under the applied field with the same mobility. However as they pass through the separating gel the proteins separate, owing to the molecular sieving properties of the gel. Quite simply, the smaller the proteins the more easily it can pass through the pores of the gel, whereas larger proteins are successively retarded by frictional resistance due to the sieving effect of the gels. Being a small molecule, the bromophenol blue dye is totally unretarted and therefore indicates the electrophoresis front, i.e. the extent of migration of the dye gives an index of electrophoretic process. The dye migrates faster than all macromolecules. When the dye reaches the bottom of the gel, the current is turned off and the gel is removed from in-between the glass plates and shaken in an appropriate stain solution. A typical gel would take 1 to 1.5 h to prepare and set, 3 h to run at 30 mA. Vertical slab gels are invariably run, since this allows up to 20 different samples to be loaded on to a single gel.
Detection, estimation and recovery of proteins in gels
Gels from a column are removed by forcing water from a hypodermic syringe around the walls of the column, allowing the gels to be extracted under gentle pressure. Slab gels are removed by introducing a thin metal plate (spatula) between the two gel plates and coaxing the plates apart. Before staining, the gels may be immersed in a fixative (5% TCA) to guard against diffusion of separated component (prevent diffusion). The most commonly used general protein stain for detecting proteins on gels is the trimethylamine sulfate dye Coomassie Brilliant Blue R-250 (CBB).
Coomassie Brilliant Blue R 250 (CBB)
Proteins are often visualized by staining with the dye Coomassie Brilliant Blue R 250. Staining is usually carried out using 0.25% (w/v) CBB in methanol: acetic acid: water (50:10:40, by volume). This acidmethanol mixture acts as a denaturant to precipitate or fix the protein in the gel, which prevents the protein from being washed out while it is being stained. Staining of most gels is accomplished in about 2 h and destaining is achieved by gentle agitation in the same methanolacid- water solution but in the absence of the dye (usually overnight). The Coomassie stain is highly sensitive; a very weakly staining band on a polyacrylamide gel would correspond to about 1 μg of protein. The CBB stain is not used for staining cellulose acetate (or protein blots) because it binds to the paper. In this case, proteins are first denatured by brief immersion of the strip in 10% (v/v) trichloroacetic acid, and then immersed in a solution of a dye that does not stain the support material, for example: Procion blue, Amido black or Procion S. Although the Coomassie stain is highly sensitive, many workers require greater sensitivity and use a silver stain. Silver stains are based either on techniques developed for histology or on methods based on the photographic process. In either case, silver ions (Ag+) are reduced to metallic silver on the protein where silver is deposited to give a black or brown band. Silver stains can be used immediately after electrophoresis or alternatively, after staining with CBB. With the latter approach, the major bands on the gel can be identified with CBB and then minor bands not detected with CBB, identified using the silver stain. The silver stain is at least 100 times more sensitive than Coomassie Brilliant Blue, detecting protein down to 0.l μg amounts.
Results
Determination of purity and molecular weight of purified human IgG: To determine the purity of isolated IgG, fraction no.2 (serum samples 1, 2 and 3), were analyzed on a 12% separating gel and 6% stacking gel. Coomassie blue stained gel is shown in Figure 13. Proteins from fraction no.2 serum samples 1, 2 and 3 resolved in to 2 bands. After determining the purity of isolated IgG, the molecular weight of isolated IgG was determined by running standard protein molecular weight markers along with fraction no.2 serum samples 1 and pure human IgG. Coomassie brilliant blue R 250 stained pattern is shown in the Figure 13.
As shown in Figure 13 (lane-1), two protein bands with molecular weights of and 50 kD and 25kD corresponding to heavy (50 kD) and light chain (25 kD) of immunoglobulin molecule were observed. Similar results were observed in the case of pure human IgG. These results clearly suggest that the IgG purified from human and serum was fairly pure.
Native–PAGE (Activity staining of the enzyme HRP): While SDS–PAGE is the most frequently used gel system for studying proteins, the method is of no use if one is aiming to detect a particular protein (often an enzyme) on the basis of its biological activity, because the protein (enzyme) is denatured by the SDS–PAGE procedure. In this case it is necessary to use non-denaturing conditions. In native or buffer gels, polyacrylamide gels are again used (normally a 7.5% gel) but the SDS is absent and the proteins are not denatured prior to loading. Since all the proteins in the sample being analyzed carries their native charge at the pH of the gel (normally pH 8.7), proteins separate according to their different electrophoretic mobilities and the sieving effects of the gel. It is therefore not possible to predict the behavior of a given protein in a buffer gel but, because of the range of different charges and sizes of proteins in a given protein mixture, good resolution is achieved. The enzyme of interest can be identified by incubating the gel in an appropriate substrate solution such that a coloured product is produced at the site of the enzyme [18]. An alternative method for enzyme detection is to include the substrate in an agarose gel that is poured over the acrylamide gel and allowed to set. Diffusion and interaction of enzyme and substrate between the two gels results in colour formation at the site of the enzyme. Often, duplicate samples will be run on a gel, the gel cut in half and one half stained for activity, the other for total protein. In this way the total protein content of the sample can be analyzed and the particular band corresponding to the enzyme identified by reference to the activity stain gel.
Result: The enzyme HRP was analyzed on a slab gels consisting of 12% separating gel and 6% stacking gel. After electrophoresis, the enzyme was identified by activity staining.
Conclusion: Experiment studies shows binding of enzymes to antibodies. This involves the formation of a stable, covalent linkage between an enzyme & Antibody. During these studies we have done many trials and studies on different Proteins, Preparation of anti- Human IgG is a big task for us which we used in this experiment, Prepared anti-human IgG HRP conjugate we have used for the following experiments like Qualitative ELISA for detection of human IgG and Identification of IgG by western blotting, For this conjugation studies we carried out by Periodate oxidation method and results are noted.