Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 8
We discovered and validated medium sized apolipoprotein (a) as a marker for good myocardial collaterization. A total of 80 subjects were investigated in two serial studies: a discovery study (n=14) applying a pooling strategy to a gel and label free proteomics platform followed by a validation study (n=80) measuring apolipoprotein(a) isoforms and concentration in individual subjects. Degree of myocardial collaterization as well as apolipoprotein(a) concentration and isoform determination were performed by state-of-the-art methodologies. As apolipoprotein(a) concentration negatively correlates with isoform size (variable number of Kringle-IV type 2 repeats in human population), subjects were grouped into patients with small, medium and large apolipoprotein(a) isoforms for the statistical analysis. Among the 70 subjects with medium and large apolipoprotein(a) isoforms (>17 Kringle-IV type 2 repeats), subjects with insufficient collaterization (n=57) had a median apolipoprotein(a) concentration of 11.9 nmol/L, while patients with sufficient collaterization (n=13) had a median concentration of 31.3 nmol/L (p=0.033, Mann-Whitney U-test). Among the 52 subjects with medium sized apolipoprotein(a) isoforms (30< Kringle IV type 2 repeats >17) the difference in concentration was even more significant (13.4 vs 33.5 nmol/L, p=0.008).
%RSD: Relative Standard Deviation; apo(a): Apolipoprotein(a); CAD: Coronary Artery Disease; CFI: Collateral Flow Index; CID: Collision-induced Dissociation; FWHM: Full Width at Half Maximum (peak intensity); GlcNAc: N-acetylglucosamine; HCII: Heparin Cofactor II; IEF: Isoelectric Focusing; IPG: Immobilized pH Gradient; LACB: Beta-lactoglobulin; Lp(a): Lipoprotein(a); OGE: Off GelTM Electrophoresis; PMSS: Peptide Match Score Summation; RP-HPLC: Reversed Phase HPLC
Blood vessels connecting left and right coronary arteries are referred to as “coronary collateral vessels” or “coronary anastomoses”. In the presence of obstructive or occlusive coronary artery disease, anastomoses between different vascular regions may serve as natural bypass for blood to reach myocardial territories distal to an arterial obstruction or occlusion, thereby preventing or mitigating myocardial infarction. The therapeutic promotion of collaterals would offer an alternative treatment strategy for coronary artery disease (CAD) patients. Today, the presence of sufficient collaterals to prevent ischemia during coronary occlusion can only be determined by invasive methods. The collateral flow index (CFI) determination, using ultra thin Doppler and pressure angioplasty guide wires, is currently the only quantitative method for the determination of coronary collateral flow in human patients. It has been determined empirically that a CFI 0.25 is sufficient to prevent myocardial ischemia in the event of a one-minute coronary occlusion (Pohl et al., 2001). Furthermore it has been shown that sufficient collateral arterioles occur more frequently in the presence of stenotic lesions in coronary arteries (Urban et al., 1987; Wustmann et al., 2003). However the response of collateral development to a certain obstructive stimulus (higher flow rates and high shear stress in preexisting interconnecting arterioles) is highly variable and it is adequate only in about 1/3 of patients with CAD to prevent myocardial ischemia during occlusion (Pohl et al., 2001). Both, angiogenesis and arteriogenesis are involved in enhancing the degree of collaterization. The latter is thought of being the more important factor, as increasing the diameters of already existing arterioles contributes more to the blood supply than the formation of new capillaries. That is why any supporting remodeling agent or therapy which improves revascularization is of great interest. Most insight on the molecular biology of arteriogenesis in context with collateral growth has come from animal models. In short, increased shear stress in preexisting arterioles induces inflammation of the endothelium. The attracted mononuclear blood cells together with the endothelium then orchestrate the remodeling process, which includes cell proliferation, degradation and rebuilding of extra cellular matrix (Heil and Schaper, 2007).
Current therapeutic approaches to obstructive CAD include pharmacologic reduction of myocardial oxygen demand and mechanical restoration of blood flow to regions lacking sufficient supply. No agents stimulating arteriogenesis or angiogenesis are currently used in therapy, although some studies indicate that certain angiogenic factors do enhance collateral flow in the short term (Seiler et al., 2001; Zbinden et al., 2005), but are not applicable because most angiogenic factors are also atherogenic. Recently, a clinical study testing a 3-month physical endurance exercise program has shown to augment coronary collateral supply to healthy and stenotic coronary arteries (Zbinden et al., 2007).
In our study we searched for plasma protein biomarkers correlating with the degree of myocardial collaterization by comparing pooled plasma samples of patients with very well developed collaterals (CFI>0.25) to patients with very badly developed collaterals (CFI<0.1). The comparison was performed with our recently developed gel and label free proteomic screening platform (Stalder et al., 2008) employing immunodepletion of the twelve most abundant plasma proteins followed by protein fractionation prior to LC-MS/ MS based protein identification of the tryptically digested fractions. We used the probability based peptide match score summation (PMSS) as relative protein abundance indicator, an approach first shown by Allet and Colinge (Allet et al., 2004; Colinge et al., 2005) and adapted in our laboratory (Heller et al., 2007; Stalder et al., 2008). PMSS is based on spectrum sampling (Liu et al., 2004), and is based on the assumption that the frequency of detecting peptides of a certain protein correlates with its relative concentration. While spectrum counting counts the frequency of identified peptides, PMSS uses the quality (z-score) of the peptide identification as a weighting factor. The finding of differential apo(a) expression discovered with the proteomics study was then successfully validated by measuring specific protein concentrations in all 80 subjects.
Patient Samples
Patients undergoing coronary angiography because of chest pain or a positive treadmill exercise test were enrolled for this study after written consent of the patients and with approval of the local ethical committee. Blood samples of 80 patients (age 61±11 years, 15 women) referred for diagnostic coronary angiography and undergoing invasive collateral assessment were drawn into EDTA containing tubes. Clinical parameters of all subjects are given in Appendix C and no statistical significance was apparent between the two groups with sufficient or non-sufficient collateralization based on a two-tailed students t-test assuming equal variance. After blood cells were depleted by centrifugation at 2,000 g (20°C) for 20 minutes, protease inhibitor cocktail CompleteTM from Roche was added according to manufacturers recommendations. Aliquots of 250μL and 1 mL were stored at -80°C. After all samples were collected, seven aliquots of patients with very high and seven patients with very low collateral flow index (CFI>0.25: Pool A, CFI<0.1: Pool B) were thawed at 37°C for 5 minutes, pooled into two pools, divided into 100 μL aliquots and stored at -80°C.
Assessment of Collateral Flow
Invasive assessment of coronary collateral flow index was performed by simultaneous measurement of distal coronary occlusive pressure (Poccl), aortic pressure (Pao) and central venous pressure (CVP) during a 1-minute coronary balloon occlusion. CFI was calculated with the following formula: CFI=( Poccl-CVP)/( Pao-CVP) (Seiler et al., 1998).
Fractionation of Plasma Proteins
Plasma pool samples (100 μL) were thawed at 37°C for 5 minutes and 32 μL of plasma was depleted of the 12 most abundant plasma proteins (Albumin, IgG, Transferrin, Haptoglobin, Alpha-1-Antitrypsin, Alpha-2-Macroglobulin, IgA, IgM, Orosomucoid, ApoA-I, ApoA-II, Fibrinogen) through immunodepletion using SepproTM IgY-12 depletion kit (GenWay, San Diego, CA, USA) according to the provided standard protocol. Depleted plasma proteins (~250 μg) were immediately fractionated into 51 fractions according to their hydrophobicity by RP-HPLC on a Summit HPLC System (Dionex, Idstein, Germany) using a GRACE VIDAC C4 column (4.5 mm i.d. x 150 mm length), which was kept at 58°C, and a Foxy fraction collector under the control of Chromeleon Software (Dionex, Idstein, Germany). The samples were acidified by addition of TFA to the final concentration of 0.1% (v/v), incubated, centrifuged and loaded onto the column by two consecutive injections of 200 microL at a flow rate of 0.75 mL/min of solvent A (0.1% TFA in water/acetonitrile 97:3). Columns were developed by a sigmoid acetonitrile gradient of 3 to 100% solvent B (0.1% TFA in water/acetonitrile 3:97) in 54 min at a flow rate of 0.75 mL/min. Fractions of 0.75 mL were collected each minute. The UV trace at 214 nm and 280 nm as well as the back pressure of the column was recorded by the software. Fractions were collected in 1.5 mL Eppendorf tubes which were closed, labeled, and pierced in the cap with a needle before storage at -80°C. The frozen fractions were then lyophilized overnight in a Christ Alpha 2-4 LSC freeze dryer (Kühner AG, Birsfelden, Switzerland). The lyophilized protein fractions were kept at -20°C until further use.
Protein Digestion, LC-MS/MS and Protein Identification
Sequencing grade modified trypsin was obtained from Promega (Catalys, Wallisellen, Switzerland). Formic acid, LC-MS grade acetonitrile, Tris buffer and ammonium bicarbonate, were purchased from Fluka (Buchs, Switzerland). Lyophilized RP-HPLC samples were reconstituted and digested by adding 14 microL of 0.1 M Tris pH 8, 5 mM DTT, 50 ng/microL trypsin and snapping the tube with the index finger, in order to dissolve all of the lyophilized proteins. The liquid was spun down and incubated overnight at room temperature. In order to maximize reproducibility, proteins were not alkylated prior to digestion, but constantly kept in a reducing environment. The digestions were stopped by adding 2 microL of concentrated formic acid. Digests were analyzed by LC-MS/MS by loading 95% of the sample on a homemade 0.15 x 50 mm C8 RP column, applying a 40-minutes acetonitrile gradient on an Esquire3000 ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany) essentially as described elsewhere (Heller et al., 2007).
Peak lists from the raw data were created by Data Analysis version 3.1 (Bruker Daltonics, Bremen, Germany) using the following parameters. MS/MS compounds exceeding a total ion chromatogram intensity of 4000 ion counts were exported and all spectra from the same precursor eluting within a retention time window of 0.5 minutes were compiled to one MS/MS peak list. MS/MS peak detection was made with the Apex peak finder algorithm using a peak width at half maximum (FWHM) of 0.1 m/z, a signal-to-noise ratio (S/N) of one, a relative to base peak intensity threshold of 2%, and an absolute intensity threshold of 10 ion counts as parameters. A mixed list of deconvoluted and nondeconvoluted MS and MS/MS signals, with an allowance for only the 200 most abundant peaks from non-deconvoluted MS/MS signals of each spectrum, were exported into Mascot generic file format text (mgf) files. MS signal deconvolution was set to “Auto” for resolved isotope, and a maximum charge of four with minimally three peaks in set and a molecular weight agreement of 0.05% for related ion deconvolution, respectively. MS/MS peak deconvolutions were allowed for a maximum charge of one only. S/N and FWHM values were also exported into the mgf files.
CID spectra interpretation was performed with the PHENYX v2.1 search engine on the vital-it.ch server operated by GeneBio (Geneva, Switzerland) against the latest human UniProt-SwissProt 51.0 (295981 individual entries) protein database allowing variable modifications of Met oxidation, Asn/Gln deamidation and formylation of free amino groups. Parent and fragment mass tolerances were set to 2 and 0.8 Da. Up to two missed cleavages and half tryptic peptides were allowed. Peptide identifications with z-scores>8 were accepted, but reconsidered if a protein identification was based on a single peptide. Each CID spectrum was allocated to only one peptide identification, thus preventing identification redundancy.
Protein Identification Acceptance Criteria
False positive rates (FPR) were determined by applying five representative vivid peak lists from different regions of the fractionations to the human UniProt-SwissProt 51.0 (295981 individual entries) database and to the reversed decoy database of equal size. The FPR are shown in appendix A. A FPR of zero was observed with z-scores above 9.2, which corresponds to p-value <1016. A false positive rate of less than 3% was observed at z-scores >8. Thus protein identifications were automatically accepted when at least two different peptides with z-scores >8 were found (FPR: 0.032). Protein identifications were considered reproduced when found in identical fractions across all runs. A shift of one RP-HPLC fraction was tolerated.
Data Processing and Peptide Match Score Summation (PMSS)
Several Perl scripts were developed and used in this study. They are available upon request from the authors. We hereby offer a short description of the dataflow and the involved scripts:CID spectra interpretation was performed on the PHENYX web server, which is based on the probabilistic OLAV scoring algorithm (Colinge et al., 2003; Magnin et al., 2004; Colinge et al., 2004). The resulting xml files (one result file per protein fraction) containing peptide identifications and their probabilistic identification scores, were downloaded from the server. Semi quantitative protein abundance scores and abundance distributions were calculated within individual fractions and the complete analysis by summing peptide match scores (z-scores >7), a technique called peptide match score summation (PMSS) (Colinge et al ., 2005; Heller et al ., 2007). This resulted in comparable PMSS based protein abundance scores and abundance distribution patterns for every protein identification. Some details: In case a peptide could be assigned to two different protein identifications, the score of the peptide was only added to the one with the higher sequence coverage. If a peptide was identified several times, all z-scores were added to the PMSS score, as long as it was derived from a different MS/MS spectrum. Peptide identification acceptance criteria (zscores>8) were more stringent than criteria allowing peptides to contribute to PMSS based abundance scores (zscores>7). The Phenyx z-score is a true probabilistic score without bias by peptide length or charge. Z-score based PMSS abundance representation therefore correlates extremely well with the spectrum sampling approach (Colinge et al ., 2005; Heller et a ., 2007).
Apolipoprotein(a) Size and Molar Lp(a) Concentration Determinations
The levels of Lp(a) in plasma were measured in all 80 subjects by a double monoclonal antibody-based ELISA. The detecting monoclonal antibody in this assay is specific to a unique epitope in apo(a), namely Kringle 4 (K4) type 9 and therefore, the accuracy of the assay is not affected by apo(a) size polymorphism (Marcovina et a l., 1995). Lp(a) concentrations were reported in nmol/L. The apo(a) isoform size was determined in all 80 subjects by high-resolution sodium dodecyl sulfate-agarose gel electrophoreses followed by immunoblotting as previously reported (Marcovina et al., 1993). Briefly, individuals with two apo(a) isoforms well defined in the gel were immediately assigned the relative kringle number to each isoform. Analysis of all samples with only one apparent isoform were repeated using an increased amount of plasma to verify whether or not it was possible to detect a second isoform present in a very small amount. Samples with no clear separation between two closely migrating bands were also reanalyzed. It has been determined that the logarithm of the number of K4 obtained by genotyping is highly correlated with the mobility of the isoforms on agarose gel (Marcovina et al., 1996b). Therefore, the apo(a) isoforms are designated by the relative number of Kringle 4 type 2 domains.
Statistical Analysis
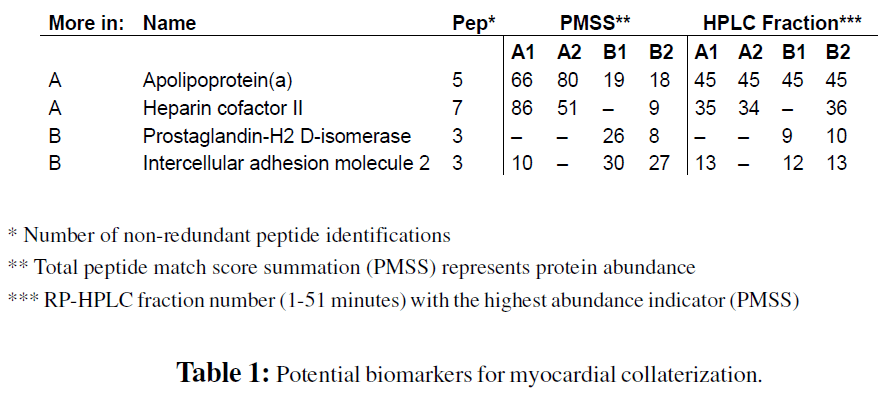
For the relative protein abundance assessment from the proteomics screen, a random ratio analysis (Stalder et al., 2008) using PMSS values of protein identification was performed. Proteins standing out from the normal noise level were considered as potential biomarkers for myocardial collateralization (Table 1). Due to reasons of availability only concentration levels of apo(a) was chosen for validation and measured in all 80 patient samples by immuno-affinity methods and tested for significant differences in median protein concentrations between patients with sufficient and insufficient collateral flow index by the non-parametrical Mann-Whitney U-test.
Proteome Screening of Plasma Pools
The comparative protein profile analysis of the two pools was performed twice, which allowed for a rough estimation of the reproducibility of the comparison. Furthermore the two pooled samples were compared in a preliminary analysis using a different RP-HPLC column (mRP-C18, Agilent Technologies, Waldbronn, Germany). The results are not shown, but were used to confirm the results from the presented screen. The PMSS based abundance profiles of protein identifications were well reproduced in the four runs, with abundance profiles from replicates matching better than the ones from differing pool samples (Figure 1). In the four runs a total of 1601 different peptides were identified, whereof 1505 possessed one or more sibling peptide describing the same parent protein. In total, 121 different proteins were identified by at least two non-redundant peptides.
Figure 1: PMSS profiles of proteins identified in all four samples. This figure demonstrates the high reproducibility of the profiling platform and gives an impression over the fractionation power of the system. Each line on the y-axis represents an identified protein of a non redundant protein list, which was sorted by fraction of maximal identification, whereas white pixels stand for high PMSS scores.
The additional 94 one peptide protein identifications (all with a z-score >8) were not used for the screen, but might still be of interest with respect to the characterization of the human plasma proteome. From the 121 different protein identifications based on two or more peptides, 84 identifications were found in all four runs and nine identifications were only found with pool A (CFI>0.25), six proteins were found solely in pool B (CFI<0.1) and eight identifications were not reproduced in pool A or pool B, despite possessing two peptide identifications (Appendix B). Furthermore fourteen protein identifications could be accredited to keratin and IgG chains. A non-redundant list of peptide identifications is shown in Appendix A and a non-redundant list of all protein identifications and their four varying abundance indicators, the peptide match score summations (PMSS), are shown in Appendix B.
Spectrum sampling methods, like PMSS, as well as peak area based approaches require several reproducible peptide identifications per protein in order to produce accurate abundance ratios, thus no significant statements can be made of the numerous protein identifications near the detection limit. Therefore, we considered protein identifications as potential markers discriminating sufficient from poor CFI when one of the following conditions was observed: 1) protein identifications based on at least two peptides found in both samples of one pool but not in the samples of the other, 2) protein identifications found in both samples of both pools with strongly differing abundance indicators between the samples of the two; 3) qualitative difference represented by differing abundance distribution based on HPLC retention time. Furthermore, all findings from the doubly performed comparison had to be confirmed by the third stand alone comparison using a different RP-HPLC column. Protein identifications which met those conditions and were considered for validation are listed in Table 1 and the underlying peptide identifications are listed in Table 2.

The three times higher abundance indicator of apolipoprotein(a) in the pooled samples of patients with very good myocardial collaterization represents the most solid result of the screen, mainly because it was identified in all three comparative studies and because the abundance indicator ratios were very well reproduced. One apolipoprotein(a) protein is covalently linked to one LDL molecule via ApoB-100 through a single disulfide bond connecting their C-terminal regions. This dimeric macro protein forms lipoprotein(a) (Lp(a)), a lipid transporting particle in plasma. ApoB-100 was also identified in the HPLC fractions 44 and 45 where apo(a) was detected. However, ApoB-100 exists in many isoforms of variable molecular size. Consistently, ApoB-100 eluted as a dispersed peak from HPLC fraction 42 to the end of fractionation therefore confounding the real ratio of Lp(a) derived ApoB-100. Thus, it was not possible to confirm the apo(a) ratio with the ApoB- 100 ratio.
Lp(a) is a very interesting particle for several reasons. First of all its physiological function is unknown but as it independently evolved in primates and hedgehogs it must have a specific function. Secondly, Lp(a) is known as risk factor for cardiovascular disease and has shown atherogenic properties. As atherogenesis and arteriogenesis, which is responsible for collateral growth, are based on similar mechanisms, it is conceivable that apo(a) is involved in the promotion of atherosclerosis and arteriogenesis. The high heterogeneity in protein size (300-800 kDa) in the human population makes apo(a) a very special protein. The heterogeneity is based on a varying number of kringle 4 type 2 (K4 type 2) repeats (2-40) coding region in the apolipoprotein(a) gene. This large variation was caused by neutral evolution in the absence of any selection advantage. It has been shown, that individuals with short apo(a) isoforms have higher concentrations of lipoprotein(a) and a higher risk for CAD or preclinical vascular changes (Berglund et al., 2004). On the other hand, elevated Lp(a) levels have been found in centenarians (Thillet et al., 1998). It has been shown that apo(a), especially the variable K4 type 9 repeats at the N-terminus of the protein, may be physiologically operative in modulating angiogenesis, a process involved in the early formation of collateral vessels (Morishita et al., 2001; Schulter et al., 2001). Furthermore apo(a) competitively inhibits fibrinolysis due to its homology to plasminogen (Hajjar et al., 1989) and contributions to plaque and stenosis formation due to interference with transforming growth factor-b activation (Grainger et al., 1994). It is also worth mentioning that Lp(a) levels are particularly affected by apo(a) synthetic rate, which is subject to strong genetic regulation. Thus Lp(a) plasma levels are affected only to a minor extent by age, sex and environmental factors (Utermann, 1989). However, there is a published study which showed that the serum level of apo(a) is inversely associated with the development of the coronary collateral circulation (Aras et al., 2006), which completely contradicts our findings.
Apolipoprotein(a) Isoform and Concentration Determination in All 80 Samples
Apolipoprotein(a) concentration was determined with a state-of-the-art ELISA assay, using antibodies targeting a non-repetitive section of apo(a), the only proven assay to determine isoform independent concentrations (Marcovina et al., 1995). The apo(a) size was determined with pulsedfield electrophoresis (Marcovina et al., 1996a). The immunoassays confirmed the result of our proteomics screen. The seven patients with sufficient CFI from pool A had significantly higher apo(a) concentrations than the seven patients with very poor CFI values used for pool B (97 and 46 nmol/L). Including all 80 patient samples the result was confirmed too, but was not significant when applying the Mann- Whitney-U-Test. The known negative correlation between apo(a) size and Lp(a) concentration (Rifai et al., 2004) was also detected with our samples (Figure 2).

Figure 2: Apolipoprotein(a) size and concentration in relation to sufficient and insufficient myocardial collaterization (CFI value). The obvious negative correlation between apo(a) size and concentration lead to the formation of three groups of patients (small, medium and large sized apo(a) isoforms) and their testing for differences in apo(a) concentration between patients with sufficient and insufficient CFI values.
We concluded that the strong negative correlation between apo(a) size and apo(a) concentration could overshadow the less strong correlation between apo(a) concentration and the degree of myocardial collaterization, we were testing for as described by Marcovina and colleagues testing Lp(a) concentration differences between black and white Americans (Marcovina et al., 1996b). Based on this assumption, we grouped the 80 patients according to their apo(a) size: patients with less than 18 K4 type 2 repeats, patients with 18 to 29 K4 type 2 repeats and patients with more than 29 K4 type 2 repeats. Within the group with medium sized apo(a) isoforms (52 of 80 patients) a significantly higher apo(a) concentration was found in patients with sufficient CFI levels (33.5 and 13.4 nmol/L, p=0.009, Mann-Whitney-U-Test). Even when combining the groups with medium and large sized isoforms (70 of 80 patients) the correlation between apo(a) concentration and CFI value remained significant (31.3 and 11.9 nmol/ L, p=0.033). Within the patient groups with very short and very long isoforms on the other hand, no significant correlation between apo(a) size and CFI value was observed. The complete dataset is available in Appendix C and the results are summarized in Figure 3. Receiver operating characteristic curves revealed that Lp(a) concentrations are not sensitive or specific enough to predict myocardial collaterization by its own (Figure 4). Our results are based on an opportunistic study with a limited data set. Additionally, there is a considerable variation in Lp(a) levels across individuals for a given apo(a) size. These factors might blur somewhat statistical significance. From other studies it is already known that Lp(a) has some, but still ill defined functional implications in vessel biology as reviewed by (Berglund and Ramakrishnan, 2004). One explanation being that apo(a) is only one factor in a system where many factors are needed in order to tip the balance on one side or the other, e.g. atherogenesis or arteriogenesis.

Figure 3: Box plots of Lp(a) concentration as a function of CFI. Median Lp(a) concentration, the number of patients per group (n) and the possibility of the hypothesis being confirmed by coincidence (p, Mann-Whitney-U-Test) are added in the figure. Hypothesis: Patients with sufficient CFI (>0.25) possess higher Lp(a) concentrations than patients with insufficient CFI values. A: All 80 patients B: 70 patients with medium and big sized apo(a) isoforms (K4 type 2 repeats >17) C: 52 patients with medium sized apo(a) isoforms (30< K4 type 2 repeats >17)

Figure 5: Receiver operating characteristic curves (ROC) of Lp(a) concentration as a function of CFI. ROC curves were calculated with apo(a) having more than 17 K4 type 2 repeats (left panel) and medium size with more than 17 but less than 30 repeats (right panel). The area under the curve were 0.668 and 0.771, respectively. The gray lines represent the lower and upper 95% confidence interval. ROC curves were calculated with a web-based, publicly available tool (Eng J. ROC analysis: web-based calculator for ROC curves. Baltimore: Johns Hopkins University. Available from: http://www.jrocfit.org).
Interestingly our results do not confirm a recently published study on this subject (Aras et al., 2006), in fact they are contradictory. In this study the authors did not distinguish between apo(a) isoforms and the apo(a) concentrations measurements. Especially, the apo(a) concentration determination used was dependant on apo(a) isoform size which is not very accurate in a population containing different isoforms. Furthermore, the degree of collaterization was assessed by a qualitative interpretation by echocardiograms rather than an absolute and quantitative measurement as done in our study.
Results show that the recently developed screening platform (Stalder et al., 2008) is applicable to clinical questions and is capable of delivering preliminary results. With the equipment used, the approach is limited to the ~120 most abundant proteins and thus is not capable to detect lower abundant proteins.
The screen suggests a correlation between the degree of myocardial collaterization, represented by the CFI value, and the concentration of apo(a) in blood plasma. While validating this result by determining apo(a) concentration and apo(a) size in the 80 patient samples, the negative correlation between apo(a) concentration and apo(a) size was confirmed as well as the result from the proteomics screen. We hypothesized that the strong negative correlation between apo(a) size and concentration overshadows the less strong correlation between apo(a) concentration and the degree of myocardial collaterization, thus grouped patients according to their apo(a) size isoform. Within the group of patients with medium sized apo(a) isoforms a significant difference in apo(a) concentration (33.5 and 13.4 nmol/L, p=0.009, Mann-Whitney-U-Test) between patients with sufficient and insufficient myocardial collateralization was detected. The mass spectrometry response for any substance is dependent on the amount of moles ionized. The correlation of apo(a) concentration measured by immunoaffinity means and mass spectrometry as done in our study suggests that mass spectrometric analysis of apo(a) could be a promising alternative for antibody-based apo(a) concentration determination in serum. In combination with spiking of isotopically labeled, kringle specific peptides (AQUA) it would even become possible to determine the isoform of apo(a) (Gerber et al., 2003).