Journal of Physical Chemistry & Biophysics
Open Access
ISSN: 2161-0398
ISSN: 2161-0398
Research Article - (2022)Volume 12, Issue 3
Computational chemistry has come of age with important advances in computer hardware and software over the last few decades. It has realized full partnership with theory and experiment as a tool for understanding and predicting the broad range chemical, physical, and biological behavior. Computational chemistry advances will broad and encompassing, because chemistry is so crucial to the myriad of advances in areas such as designation of Organic Photovoltaic (OPV) materials, drug design, biological sciences, and chemical manufacturing industry. The use of Computer-Aided Drug Discovery (CADD) methods in preliminary studies by leading pharmaceutical companies and research groups has helped to expedite the drug discovery and development process minimizing the costs and failures in the final stage. The application of rational drug design as an integral part of CADD delivers useful insights into the understanding of the binding affinity and molecular interaction between target protein and ligand. Computational methods such as molecular docking, molecular dynamic simulation, structure-activity relationships, electronic properties, and pharmacophores were used in computational drug design. The OPV solar cell is a type of organic electronics that deals with conductive organic polymers for light absorption and charge transport to produce electricity from sunlight photovoltaic effect. OPV materials span a vast chemical space due to the structural versatility of their carbon based framework. Due to the challenges in synthesis and experimental characterization of these systems approaches that is involved in computational screening of OPV compounds can aid in accelerating the discovery of high efficiency materials. Computational methods such as morphological properties, chemical reactivity descriptors, band gap, quantitative structure-property relationships, emission quantum yield, and optical and electronic transport properties of OPV materials were employed in the computational OPV material design. The Suitable computational chemistry methods are Ab-initio, Semi-empirical and Density Functional theory methods available with software such as Dalton, Gaussian 09, Gamess, Avogadro and Authodock Vina. Generally, the aim of this review is to provide the enough information about application of computational chemistry for the drug design and OPV material design.
Computational methods; Computer-Aided drug discovery; Organic photovoltaic
Computational chemistry is the application of chemical, mathematical and computing skills to the solution of curious chemical problems by using computers to generate information of molecules [1]. Many computational chemistry researchers use electronic structure methods to predict molecular structures, chemical properties and even to predict the outcome of a chemical reaction [2]. It is chemistry in the computer instead of in the laboratory and use computer calculations to predict the structures, reactivities and other characteristics of molecules. However, computational chemistry is used to predict new molecules or new reactions which are later investigated experimentally [3]. Computational chemistry research can shed light on various inorganic, organic, organometallic and coordination compounds stability, structural features, spectral characteristics (UV-Visible, NMR, and IR), conformational transitions and rotation barriers of their functional groups [4]. It is applicable in designing drug and OPV materials. Computational chemistry software performs electronic structure, geometry optimizations; inter-reaction coordinates, molecular docking, thermodynamic parameters and spectroscopic studies of compounds (Figure 1) [5].

Figure 1: Applications of computational chemistry.
Computational chemistry insights for drug designing
Drug development is one of the most important processes in the pharmaceutical industry. Chemists have prior knowledge about the structure and desired properties of drugs before experimental synthesis by using computer. Computational drug discovery can help in identifying potential drug molecules and targets via bioinformatics [6]. In addition, it is used to assess the target structures for possible binding or active sites, generate active drug molecules, check for their dynamic and kinetic properties, the docking studies and pharmacophore site studies of drug molecules with the target molecules will help to know the affinity and efficacy of developed molecule. It is possible to rank them according to their binding affinities. Different computational methods were used in the literature review for the new drug design discovery [7].
Molecular docking
Molecular docking is currently employed computational method help to rationalize ligand activity towards a target of interest and to perform structure based virtual screening. It is a preferred method to forecast orientation of one molecule to a second, when the one molecule binds with the second molecule to form a stable complex structure. Modelling stable molecular interactions between drug molecules and target receptor molecules, ligand conformations and the most suitable ones are selected by molecular docking. Docking enables the identification of unique compounds of therapeutic interest, predicting ligand target interaction at a molecular level (Figure 2) [8].

Figure 2: Molecular docking ligand with protein.
Quantitative Structure Activity Relationship (QSAR)
Quantitative Structure Activity Relationship (QSAR) is a quantitative study of the interactions between small organic molecules and biological macromolecules. Structure activity relationships, study is significant in drug discovery to guide the synthesis of desirable new compounds as well as further characterize existing molecules. It is an approach designed to find relationships between chemical structure and biological activity or target property of study compounds. Geometrical and electronic descriptors are used to build a QSAR, which results in a mathematical model able to predict the biological activity of new compounds (Figure 3) [7].

Figure 3: Structure activity relationship of compounds by tuning substituents.
Pharmacophore mapping study
The Pharmacophore mapping study is one of the computational methods significant for description of molecular features which are essential for molecular identification and recognition of a ligand and biological macromolecule [9]. Typical pharmacophoric properties features of the compound include hydrophobic, hydrophilic, aromatic rings, hydrogen bond acceptors, hydrogen bond donors, positive charge and negative charge. This method uses the chemical features of most active and inactive compounds along with their biological activity (Figure 4) [10].

Figure 4: Pharmacophore mapping study of computational methods.
Computational chemistry insights to OPV material designing
The organic photovoltaic solar cell is a kind of organic electronics that deals with conductive organic polymers for light absorption and charge transport to produce electricity from sunlight. Among current solar cell design paradigms, organic photovoltaic cell technology shows significant potential due to its potential low cost, flexibility, and manipulability [11]. The chemistry of materials clearly understood the critical role of computations to aid materials-by-design, even as larger initiatives have been advanced. Enhanced computational innovation of high performance materials for organic photovoltaic by means of cheminformatics was studied [12].
Structural properties
Quantum chemical methods are used to study the series of compounds to establish the structure-efficiency relationship. Thanks to computational methods, that it is possible to study large numbers of compounds even before synthesis. Structure efficiency relationship has been developed for a variety of applications, which include single-molecule, intermolecular and reactive properties [13]. The study of geometric structures, electronic, optical and vibrational properties of the compounds by theoretical calculations can promote reasonable interpretation of experimental results and subsequently better understanding of the relationship between the structure and resulting properties [14]. From the optimized structure of OPV compounds, geometrical parameters such as bond length, bond angle and dihedral angles can be investigated [15].
Band gap analysis
Several researches focused on practical applications including electroluminescence displays, photovoltaic devices, thin film transistors, and so on. Analysis of the performance of narrowband gap organic solar cells was studied [16]. In recent years, many investigations on the design and synthesis of low-band gap electrochromic polymers have been the major attractive area and a most successful and flexible strategy to design small band gap polymers involves the alternation of electron-rich and electron deficient units along the polymer chain [15]. In many researches, computational methods applied successfully to predict detailed morphological information for known conjugated organic molecules, such as molecular geometries, electronic structure, Frontier Molecular Orbital (FMO) energy levels, absorption spectra, and Intramolecular Charge Transfer (ICT). Solar cell efficiencies are closely related to FMO energies and band gaps (Eg) [17]. Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) energetic levels were interpreted as the valence band (HOMO) and the conduction band (LUMO), in terms of band theory. The energetic differences between these two levels were readily interpreted as the band gap energy, which is a representative signature found on OPV materials (Figure 5) [18].

Figure 5: Band gap analysis of OPV materials.
Electron transport properties
Organic Photovoltaic materials contain two active materials: Electron acceptors and electron donors [19]. The simplest multilayer OLED consists of an anode, an Electron Transporting Material (ETM) layer, a Hole Transporting Material (HTM) layer, and a cathode [20]. One of the key challenges on the path to developing high performance OPV material is the design of a stable HTM layer that would be characterized by a high efficiency of hole injection and adequate hole mobility.
The charge transport balance is crucial for optoelectronic materials. Therefore, it is important to investigate their Ionization Potentials (IPs), Electron Affinities (EAs) and reorganization energies (l) to evaluate the energy barrier for injection and transport rates of the holes and electrons [21]. The device performance of OLEDs depends on the charge injection, transfer and balance as well as the exciton confinement Computational study improvement of organic materials photophysical capabilities of a new Hole Transporting Material (HTM) and Electron Transporting Material (ETM) which constitute the photoactive components in OPV cell device can increase the performance of such solar energy conversion materials (Figure 6) [19].

Figure 6: Design of an OSC (a): (1) Incident photons, (2) Exciton formation, (3) Exciton diffusion, (4) Exciton dissociation, (5) Charge transport, and (6) Collection. (b): Configuration of the electron-donor material and the sketched PCBM structure.
Emission quantum yield
The emission quantum yield (Φ) is a measure of efficiency of photon emission as defined by the ratio of the number of photons emitted to the number of photons absorbed. The emission quantum yield (Φ) can be affected by the competition between radiative and non-radiative decay rate constants, which can be generally formulated as [21]. The non-radiative processes associated with molecular rotations and vibrations are inactive and relatively high fluorescence quantum yields can be achieved (Figure 7).
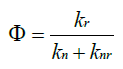

Figure 7: Emission quantum yield analysis of OPV materials.
Non-linear optical studies
Non-linear optical properties are very important properties and they find applications in the discovery of novel organic electronic materials like OLED, organic transistors, organic semi-conductors etc. [22]. Polarizability and hyperpolarizability offer useful information for frequency changing, optical modulation, optical switching and optical logic for technologies evolving in areas such as NLO activity, communication, signal processing, and optical interconnection. Organic materials are expected to have relatively strong NLO properties, due to the delocalized electrons in the π->π* orbitals [23].
Objective of the review
The investment in new drug development has grown significantly in the past decades; the output is not positively proportional to the investment because of the low efficiency and high failure rate in drug discovery. Consequently, various approaches have been developed to shorten the research cycle and reduce the expense and risk of failure for drug discovery. CADD is one of the most effective methods for reaching these goals [24]. However, the relationships between material properties, device configuration, and performance are complex and often poorly understood. Computational chemistry can calculate several important properties of photovoltaic materials that affect efficiency.
Computational calculations are easy to perform, safe, less costly, performed on any chemical system when it compared with experimental analysis. The main aim of computational chemistry is to understand the course and extent of chemical phenomena in a condition that are difficult or even impossible to observe directly in the laboratory [25]. Semi-empirical, Hartree-Fock and DFT are common and they are available in Gaussian software. GAMESS and DMOL software are available in quantum chemical calculations. Among them, Gaussian software is the most applicable one [26].
Ab-initio methods (Hartree-Fock methods)
Hartree-Fock method is the simplest kind of Ab-initio calculation. This method breaks many electron Schrödinger equations into many simpler one electron equations. Each one electron equation is solved to yield a single electron wave function called an orbital energy. The orbital describes the behavior of an electron in the net field of all the other electrons. The limitation of HF calculation is that they do not include electron correlation. It takes into account the average effect of electron repulsion, but not the explicit electron-electron interaction. Within HF, the probability of finding an electron at some location around an atom is determined by the distance from the nucleus but not the distance to the other electrons. Therefore, it can be improved by adding determinants and generating solutions which can be made to converge to the exact solution of electronic Schrödinger equation [27].
Semi-empirical methods
Semi-empirical calculations are based on approximate solutions of the Schrödinger equation with appeal to fitting to experiments. However, more approximations are made in solving it and the program draws on a kind of library of integrals that was compiled by finding the best fit of some calculated entity like geometry or energy to experimental values. This plugging experimental values into a mathematical procedure to get the best calculated values is called parameterization. Theory and experiment mixed that makes the method “semi-empirical method” [25]. It is much faster than the Ab-initio calculations, but semi-empirical calculation results can be slightly defective. There are a variety of semi-empirical methods. Among the best known are AM1, PM3 and MNDO.
Molecular mechanics
Molecular mechanics was coined in the 1970s to describe the application of classical mechanics in the determinations of molecular equilibrium structure. Chemists visualize molecules in terms of bond lengths, bond angles and dihedral angles. In the molecular mechanical methods, atoms are treated as spheres whose mass depends on the element and chemical bonds are treated as springs whose stiffness depends on which elements are bound together and whether the bond is single, double or triple. It makes no reference to electrons, and cannot throw light on electronic properties like charge distributions, neuclophilic and electrophilic behaviour [25].
Density Functional Theory (DFT) method
DFT is an electronic structured method utilized for the prediction of crystalline, clusters, molecular and atomic properties. A density functional is then used to obtain the energy for the electron density. A functional is a function of a function, in this case, the electron density. Today DFT is an important tool employed in theoretical chemistry due to its lower computational cost than Coupled Cluster methods for systems with hundreds or more atoms. DFT uses the ground state electron density instead of the wave function in order to find the ground state energy of a system by minimizing the energy functional (E[ρ]). The general form of E[ρ] contains the kinetic energy (T[ρ]), the external nuclear-electron potential (Vne[ρ]), the electron-electron interaction potential (Vee[ρ]) and the nucleus-nucleus repulsion potential (Vnn) [26].
Molecular dynamics method
Molecular dynamics consist of examining the time dependent behavior of a molecule, such as vibrational motion or Brownian motion. This is most often done within a classical mechanical description similar to a molecular mechanics calculation. The application of molecular dynamics of solvent/solute systems allows the computation of properties such as diffusion coefficients or radial distribution functions for use in statistical mechanical treatments. Usually the scheme of a solvent/solute calculation is that a number of molecules (perhaps 1000) are given some initial position and velocity. New positions are calculated a small time later based on this movement and this process is iterated for thousands of steps in order to bring the system to equilibrium and give a good statistical description of the radial distribution function (Figure 8) [28].

Figure 8: Relation between computational methods.
Computational chemistry software
Many self-sufficient computational chemistry software packages exist. Some include many methods covering a wide range, while others concentrate on a very specific range. Details of most of them can be found in: Bio-molecular modelling programs, Molecular mechanics programs, Molecular design software, Semi-empirical programs (Table 1) [29].
| Name of software | Use |
|---|---|
| Gaussian 09 | To predict energies, molecular structures, spectroscopic data (NMR, IR, UV, etc.) and many more advanced calculations. |
| GAMESS (General Atomic And Molecular Electronic Structure System) | To understand the potential of a drug molecule binding with proteins for drug discovery. |
| Chemcraft | Graphical visualization program for quantum chemistry computations. |
| Hyperchem 7.5 | Sophisticated molecular modeling environment for its quality, flexibility, and ease of use. |
| AMBER (Assisted Model Building And Energy Refinement) | It is a suite of Biomolecular simulation programs. |
| GROMOS (GROningen Molecular Simulation), | It is the name of a force field for molecular dynamics simulation, and a related computer software package. |
| GROMACS (GROningen MAchine for Chemical Simulation) | It is a molecular dynamics package mainly designed for simulations of proteins, lipids, and nucleic acids. |
| NAMD (Nanoscale Molecular Dynamics) | It is noted for its parallel efficiency and is often used to simulate large systems. |
| AUTODOCK (Automated docking) | It is especially effective for protein-ligand docking. |
| SPARTAN | It is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, Ab-initio models, density functional models, post-Hartree-Fock models, and thermochemicals. |
| PyMOL (Proprietary molecular visualization) | It is a user-sponsored molecular visualization system on an open-source foundation, maintained and distributed by Schrödinger. |
| AVOGADRO | It is a molecule editor and visualizer designed for cross-platform use in computational chemistry, molecular modeling, Bioinformatics, materials science, and related areas. |
| Crystal Explorer | It is designed for analysis the crystal structure. |
Table 1: Computational chemistry softwares and their uses.
In this review, the application of computational chemistry in drug design and organic photovoltaic material design has been reviewed. Computational chemistry is a branch of chemistry that uses computer simulation to assist in solving chemical problems. It uses methods of theoretical chemistry, incorporated into computer programs, to calculate the structures and properties of molecules, groups of molecules, and solids. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. In drug design and discovery, diverse computational chemistry approaches are used to calculate and predict events, such as the drug binding to its target and the chemical properties for designing potential new drugs. In addition, it is used to assess the target structures for possible binding or active sites, generate active drug molecules, check for their dynamic and kinetic properties, the docking studies and Pharmacophore site studies of drug molecules with the target molecules will help to know the affinity and efficacy of developed molecule. In addition to this, the organic photovoltaic solar cell is a kind of organic electronics that deals with conductive organic polymers for light absorption and charge transport to produce electricity from sunlight. The chemistry of materials clearly understood the critical role of computations to aid materials-by-design, even as larger initiatives have been advanced.
To get high efficiency OPV materials, structural Properties, band gap analysis, emission quantum yield, Non-linear optical analysis, electron transport properties of computational chemistry is important. Computational calculations are easy to perform, safe, less costly, performed on any chemical system when it compared with experimental analysis. The Suitable computational chemistry methods are Ab-initio, Semi-empirical and Density Functional theory methods available with software such as Dalton, Gaussian 09, Gamess, Avogadro and Authodock Vina. Generally, the aim of this review is to provide the enough information about application of computational chemistry for the drug design and OPV material design.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Tegegn DF (2022) Application of Computational Chemistry in Drug Design and Organic Photovoltaics Material Design. J Phys Chem Biophys. 12:330.
Received: 01-Jun-2022, Manuscript No. JPCB-22-17692; Editor assigned: 03-Jun-2022, Pre QC No. JPCB-22-17692 (PQ); Reviewed: 17-Jun-2022, QC No. JPCB-22-17692; Revised: 24-Jun-2022, Manuscript No. JPCB-22-17692 (R); Published: 01-Jul-2022 , DOI: 10.35248/2161-0398.22.12.330
Copyright: © 2022 Tegegn DF. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.