Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 7
MicroRNAs (miRNAs) are a class of small non-protein-coding RNAs that play important regulatory roles by targeting for cleavage or translational repression and involved in diverse biological functions. Accumulation of large amount of biological data indicates that miRNAs can function as tumor suppressors and oncogenes. Mutation, misexpression, and altered mature miRNA processing are implicated in carcinogenesis and tumor progression. Common single-nucleotide polymorphisms (SNPs) in miRNAs may change their property through altering miRNA expression and/or maturation, and thus they may have an effect on thousands of target mRNAs, resulting in diverse functional consequences. In this work we used computational tools to predict the functional role of mRNAs targeted by miRNA in colon cancer genes. We have presented a method which allows the use of PupaSuite, UTRscan and miRBase as a pipeline for the prediction of miRNA and their target, and evaluated the functional role of mRNA in colon cancer.
Keywords: miRNA; SNPs; miRBase; PupaSuite; UTRscan
Identifying the genes and mutations underlying phenotypic variation is one of the primary objectives of modern genetics, especially for traits of medical importance. Over the past decades, studies have analyzed and unveiled the genetic variants in the human genome, such as single-nucleotide polymorphisms (SNPs) (Stranger et al., 2007) which contribute to gene expression variation, and eventually to phenotypic variation in human populations (Rockman and Kruglyak, 2006). Single-nucleotide polymorphisms (SNPs) are the most frequent variation in the human genome, occurring once every several hundred base pairs throughout the genome. They have been studied extensively for defining the regions of disease candidate genes (Bernig and Chanock, 2006). The majority of SNPs in the genome occur in untranslated, intronic or intergenic regions. These SNPs could affect complex diseases through their effect on gene expression quantitatively. It is increasingly recognized that regulatory mutations could make a significant contribution to genetic variation, especially for complex traits, including disease susceptibility. Recent reports have shown that mutations in the coding region disrupting sequences recognized by splicing regulators such ESE or ESS can be considered an additional mutation mechanism leading to disease in humans (Pagani et al., 2003). This finding is particularly important for genetic counseling, where the pathogenicity assessment of any nucleotide substitution is crucial to correctly predict cancer risk. Taking in to this account, our group recently published a review on the use of computational tools to identify deleterious SNPs in both coding and noncoding regions taking TP53 as a pipeline gene (George et al., 2008).
MicroRNAs (miRNAs) are small 21-25 nucleotide non-protein-coding RNAs that comprise an evolutionarily conserved class of ribo-regulators which modulate gene expression via the RNA interference pathway (Ambros, 2004). miRNAs are thought to regulate gene expression post-transcriptionally by forming Watson-Crick base pairs with target mRNAs. miRNAs use two distinct post-transcriptional mechanisms to down-regulate gene expression. They act by binding to the complementary sites on the 3' untranslated region (UTR) of the target gene to induce cleavage with near perfect complementarity or to repress productive translation (Brennecke et al., 2005) and also facilitate deadenylation, which leads to rapid mRNA decay (Wu et al., 2006). Accumulation of large amount of biological data suggests that a single miRNA could bind to hundreds of mRNA targets, and these targets could be implicated in the regulation of almost every biological process (Zamore et al., 2005). miRNAs assert their function as oncogenes or tumor suppressor genes via several potential mechanisms in various forms of cancers (Calin et al., 2002; Cimmino et al., 2005; Borkhardt et al, 2006). Literature survey shows that miRNAs are also involved in solid tumors such as lung cancer, breast cancer and colorectal cancer (CRC) (Iorio et al., 2005; Karube et al., 2005; Michael et al., 2003). Loss or amplification of miRNA genes has been reported in a variety of cancers, and altered patterns of miRNA expression may affect cell cycle and survival programs (Yanaihara et al., 2006).
Because small variation in the quantity of miRNAs may have an effect on thousands of target mRNAs and result in diverse functional con-sequences, the most common genetic variation, SNPs, in miRNA sequences may also be functional and therefore may represent ideal candidate biomarkers for cancer prognosis. In recent years, few studies have described role of naturally occurring human polymorphisms associated with miRNAs and their target sites (Iwai et al., 2005; Chen and Rajewsky, 2006). Clop et al., 2006 stated that the SNP may affect organismal phenotype by altering a miRNA target site, leading to a significant alteration of protein expression. Given the wealth of data that is currently available in databases for human SNPs, such as the human genome variation database, HGVBase (Fredman et al., 2002) and the National Center for Biotechnology Information (NCBI) database, dbSNP (Smigielski et al., 2000), we can begin to identify naturally occurring variation associated with miRNAs and their targets using an in silico approach. SNPs in critical components of the miRNA system may have important phenotypic consequences, with implications for both evolutionary studies and biomedical research. In this study, we conducted a bioinformatics genome-wide survey on genes causing colon cancer and identified miRNA and their target mRNA using computational algorithims like PupaSuite (Reumers et al., 2008) which uses miRanda (John et al., 2005) and miRBase (Griffiths-jones et al., 2008). We searched for SNPs that would potentially affect novel target sites in humans and speculate that some of these variations may have functional effects using UTRscan and PupaSuite. These methods are based on different principles and merging them into a pipeline for predicting the function of target mRNA is therefore meaningful. Flow-chart of the proposed methodology is depicted in Figure 1.

Figure 1: Flow-chart of the proposed methodology for the prediction of miRNA and their targets.
Searching of Gene Ids and their Single Nucleotide Polymorphisms
A total of 20 genes responsible for causing colon cancer (Familial adenomatous polyposis and Lynch syndrome) as shown in Table 1 were collected from OMIM and HGMD and submitted to PupaSuite to extract miRNA and their target mRNA. Based on the results obtained from PupaSuite, we retrieved the information for UTR SNPs of the genes namely APC, AXIN2, CTNNB1, MAP2K4, MLH1, MSH2, MSH3, MYH, TP53 and STK11 from the Human genome variation database, HGVBase and National Center for Biotechnology Information (NCBI) database, dbSNP for our computational analysis.

Scanning for miRNA and their Target Sites
PupaSuite is a unique and more integrated interface of PupaSNP (Conde et al., 2004) and PupasView (Conde et al., 2005) are now synchronized to deliver annotations for both noncoding and coding SNP, as well as annotations for the SwissProt set of human disease mutations. In this approach, the input consists of a list of genes (genes belonging to a given pathway, involved in a particular biological function, etc.) and the user must specify the type of gene identifiers by selecting either Ensembl or an external database (which include GenBank, Swissprot/TrEMBL and other gene ids supported by Ensembl). Pupasuite retrieves SNPs that could affect conserved regions that the cellular machinery uses for the correct processing of genes (intron/ exon boundaries or exonic splicing enhancers) and uses miRanda an algorithm for the detection of potential microRNA target sites in genomic sequences.
Analyzing miRNA and their Target Sites
For the detection of potential microRNA target sites in genomic sequences, we applied miRBase to validate our prediction. miRBase scans one or more miRNA sequences against all sequences and potential target sites are reported.
A dynamic programming local alignment is carried out between the query miRNA sequence and the reference sequence. The alignment procedure scores are based on sequence complementarity between A:U and G:C matches and not on sequence identity. Then, it takes the high-scoring alignments of those above as a score threshold, detected from local alignment and estimates the thermodynamic stability of RNA duplexes based on these alignments. For this method, miRanda utilizes folding routines from the RNAlib library, which is part of the ViennaRNA (Wuchty et al., 1999). The free energy (ΔG) of optimal strand-strand interaction between miRNA and UTR is computed by the Vienna RNA folding routines and is a measure for the thermodynamic stability of a duplex. P-values for all target sites were calculated in miRinda based on the model proposed by Rehmsmeier et al., 2004. Base P-value is computed using distribution parameters derived from the genomic background of miRanda scores.
Scanning of Noncoding SNPs
Functional significance of each SNP in untranslated region (UTR) was determined by UTRscan (Pesole and Liuni, 1999) available at (http://www.ba.itb.cnr.it/BIG/UTRScan). UTResource, which is an internet resource of sequence analysis of 5’ and 3’ UTR of eukaryotic mRNAs which are involved in many posttranscriptional regulatory pathways that control mRNA localization, stability, and translation efficiency (Sonenberg, 1994; Nowak, 1994). Briefly, two or three sequences of each UTR SNP that have a different nucleotide at an SNP position were analyzed by UTRscan, which looks for UTR functional elements by searching through user submitted sequence data for the patterns defined in the UTR site and UTR databases. If different sequences for each UTR SNP are found to have different functional patterns, this UTR SNP is predicted to have functional significance. The internet resources for UTR analysis were UTRdb and UTRsite. UTRdb contains experimentally proven biological activity of functional patterns of UTR sequence from eukaryotic mRNAs (Pesole et al., 2002). The UTRsite has the data collected from UTRdb and also is continuously enriched with new functional patterns.
Prediction of miRNA and their Target Sites
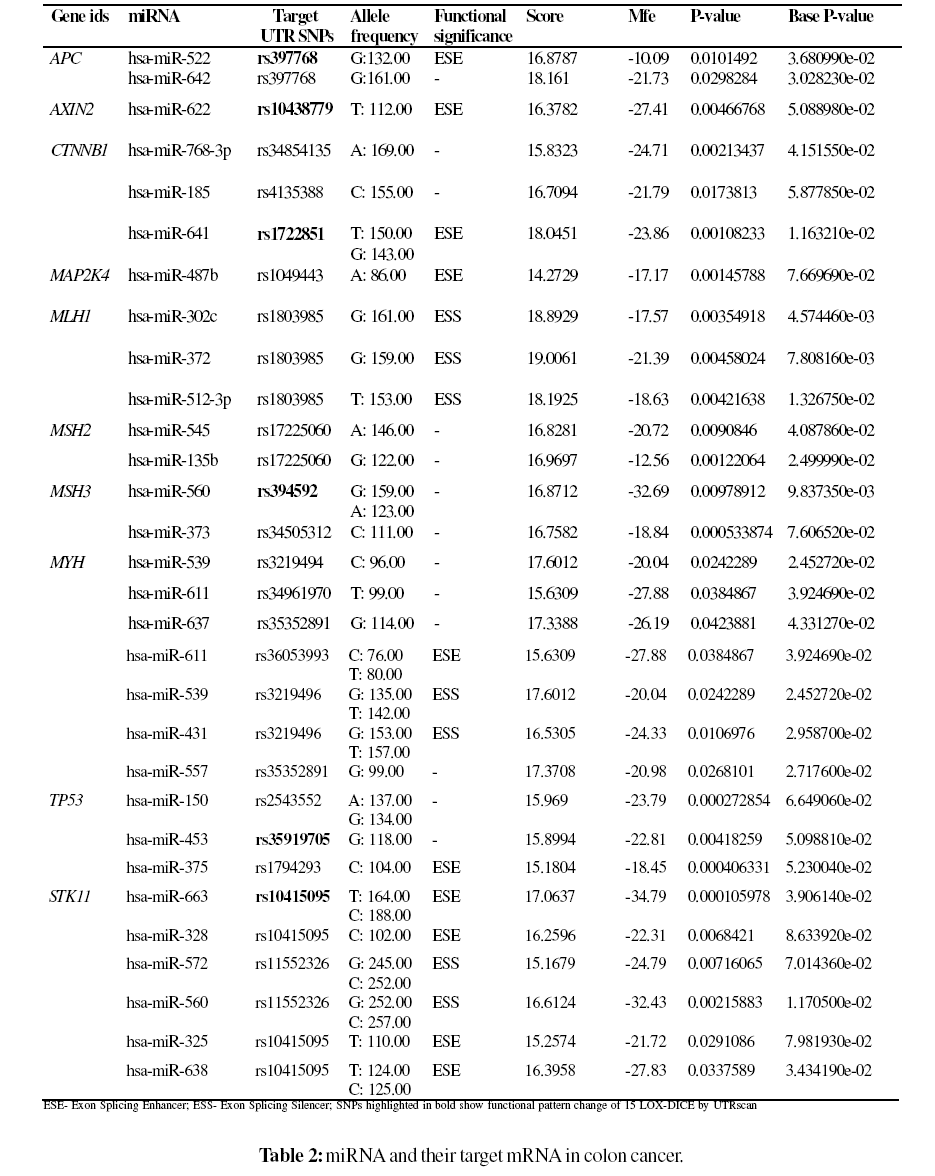
Among the 20 genes which were submitted to PupaSuite, only 10 genes (50%) displayed miRNA and their targets. Pupasuite scans the whole genome to find SNPs located at miRNAs. PupaSuite uses miRanda an algorithm for the detection of potential microRNA target sites in genomic sequences. Four miRNAs namely hsa-miR-663, hsa-miR- 328, hsa-miR-325 showed a target mRNA SNP rs10415095 and two miRNAs namely hsa-miR-638 and hsa-miR-572 showed a target mRNA SNP rs11552326 in STK11 gene, two miRNAs namely hsa-miR-637 and hsa-miR-557 showed a target mRNA SNP rs35352891 and two miRNAs namely hsa-miR-539 and hsa-miR-431 showed a target mRNA SNP rs3219496 in MYH gene, two miRNAs namely hsa-miR- 545 and hsa-miR-135b showed one target mRNA SNP rs17225060 in MSH2 gene, three miRNAs namely hsa-miR- 302c, hsa-miR-372 and hsa-miR-512-3p showed one target mRNA SNP rs1803985 in MLH1 gene and two miRNAs namely hsa-miR-522 and hsa-miR-642 showed one target mRNA SNP in APC gene as shown in Table 2. PupaSiute uses miRanda, an algorithm for the detection of potential microRNA target sites in genomic sequences, to localize all the SNPs situated in the region 3’ UTR of these targets sites. miRanda calculated the scores based on the complementarity of nucleotides (A=U or G=C) and G=U wobble pairs, which are important for the accurate detection of RNA:RNA duplexes. The result is a score (S) for each detected complementarity match between a miRNA and a potential target gene and range is between 15.1679 to 19.0061. The minimum free energy (MFE) of the miRNA– target duplex was determined while predicting the miRNA target sites. The lower MFE values of the miRNAs and the target sites reveal the energetically more probable hybridizations between the miRNAs and the target genes. It can be seen from Table 1 that miRNAs predicted by PupaSuite exhibited a low minimum free energy range from -10.09 to -34.79. It is an important noteworthy finding in miRNA and their target mRNAs analysis using PupaSuite. Recent publications suggest that multiple potential binding sites of a miRNA in a single target are good evidence for the target being regulated by the miRNA. If we consider a potential binding site being a rare event in our random model, the number of binding sites can be approximated by a Poisson distribution (Hofacker et al., 1994). The provision of P-value and Base P-value for the miRNA allows the users to assess the confidence in the prediction. Based on the above observations depicted in Table 2 the miRNAs namely hsamiR- 622, hsa-miR-641, hsa-miR-560, hsa-miR-611, hsamiR- 637, hsa-miR-557, hsa-miR-663, hsa-miR-572, hsamiR- 560 and hsa-miR-638 predicted by PupaSuite by our investigation is also well documented by on experimental protocols (Bandres et al., 2006; Cummins et al., 2006). This is an important result from this work.
Predictions of Potential Phenotypic Effect in SNPs
We further analyzed the miRNA targets and predicted the role of mRNA SNPs using PupaSuite. Besides indispensable cis regulatory motifs such as 5’ and 3’ splice sites and branch points, there are other cis regulatory sequences called exonic or intronic splicing enhancers and silencers (Fu, 2004). These sequences are recognized by a number of regulatory proteins, represented, for example, by serine/arginine- rich (SR) proteins, which bind RNA with limited sequence specificity (Liu et al., 1998). ESEs are common in alternative and constitutive exons, where they act as binding sites for Ser/Arg-rich proteins (SR proteins), a family of conserved splicing factors that participate in multiple steps of the splicing pathway (Graveley, 2000). ESSs are sequence elements that are known to regulate alternative splicing and also play a role in splice site selection (Fairbrother and Chasin, 2000).
As a result of premRNA splicing, different combinations of exons may arise and may be a ‘natural’ cause of errors in gene expression by introducing premature termination codons or altering protein structure leading to changes in spectrum of interacting proteins, intracellular localization, protein stability or posttranslational modification (Stamm et al., 2005). Seven SNPs rs397768, rs10438779, rs1722851, rs1049443, rs36053993, rs1794293 and rs10415095 with ids were predicted to disrupt the exonic splicing enhancers and three SNPs rs1803985, rs3219496 and rs11552326 with ids were predicted to disrupt the exonic splicing silencers by PupaSuite.
A revolution is underway in the approach to studying the genetic basis of cancer. In the past, most studies have focused on protein coding genes and their regulation at the transcriptional level. The recent explosion of miRNA research and discovery further underscores the importance of these regulatory molecules in many key biological processes, such as development, cellular differentiation, cell cycle control and apoptosis. Polymorphisms and mutations in the corresponding sequence space (machinery, miRNA precursors and target sites) are likely to make a significant contribution to phenotypic variation, including disease susceptibility. The mutations in miRNAs or polymorphisms in the mRNAs targeted by miRNAs may also contribute to cancer predisposition and progression (Saunders et al., 2007). Their expression profiles can be used for the classification, diagnosis, and prognosis of human malignancies. Most of the mutations identified till date lead to alter in primary sequence and hence alter in protein structure (missense or nonsense, insertion–deletions in the open reading frame, or mutations causing splicing errors). Recent studies show that regulatory mutations could make a significant contribution to genetic variation, including disease susceptibility lead to the identification of the mutations in regulatory variants (rSNPs) affecting transcript levels in cis (Pastinen et al., 2006). The most common interpretation of such cis effects is that the corresponding variants are modulating the activity of regulatory elements, including promotors and enhancers.
We applied computational tools like PupaSuite, miRBase and UTRscan to validate miRNA and their targets using colon cancer genes. PupaSuite uses miRanda algorithm for the identification of miRNAs and their target mRNA. Out of twenty genes retrieved from OMIM causing colon cancer, PupaSuite predicted miRNA and their target mRNA for only ten genes (50%). A total of thirty miRNAs and mRNAs were obtained from PuaSuite for further analysis. Of those predicted target genes in causing colon cancer (Table 2), miRNAs in genes namely APC, MLH1, MSH2, MYH and STK11 had more than one predicted target interaction site. These results suggest that 3' UTRs with more than one predicted target site for a given miRNA are more reliable than those with a single site. This concept of multiple miRNAs binding sites with target mRNA is well supported by experimental analysis in drosophila (Enright et al., 2003). Out of the predicted thirty miRNAs, ten miRNAs (33%) namely hsa-miR-622, hsa-miR-641, hsa-miR-560, hsa-miR-611, hsa-miR-637, hsa-miR-557, hsa-miR-663, hsamiR- 572, hsa-miR-560 and hsa-miR-638 predicted by PupaSuite by our investigation is also well documented by on experimental protocols (Bandres et al., 2006; Cummins et al., 2006). Functional role of target mRNA SNPs were validated using PuaSuite and UTRscan. In-silico methods provide a useful tool for an initial approach to any mutation suspected of causing aberrant RNA processing. These mutations can result in either complete skipping of the exon, retention of the intron or the introduction of a new splice site within an exon or intron. In rare cases, mutations that do not disrupt or create a splice site, activate preexisting pseudo splice sites consistent with the proposal that introns contain splicing inhibitory sequences (Baralle and Baralle, 2005). Recent studies showed that the mutations in cis splicing regulating sequences, which might shift production to mRNA with cancer-prone potential (Scholzova et al., 2007). Yang et al., 2003 showed, however, that this mutation disrupts an exonic splicing enhancer and leads to production of null protein due to aberrant splicing. Among the twenty one targeted SNPs by miRNAs, seven SNPs (38%) rs397768, rs10438779, rs1722851, rs1049443, rs36053993, rs1794293 and rs10415095 with ids were predicted to disrupt the exonic splicing enhancers and three SNPs rs1803985, rs3219496 and rs11552326 with ids were predicted to disrupt the exonic splicing silencers, whereas thirteen SNPs (62%) showed no functional significance by PupaSuite. Varied levels of alternative splicing have been detected for some of the splicing mutations in colon cancer genes (Lastella et al., 2006; Ivan et al., 2003). By UTRscan, six SNPs (29%) showed a functional pattern change 15-LOXDICE. Founding members of miRNAs were discovered by genetic screening approaches, experimental approaches were limited by their low efficiency, time consuming, and high cost. As a consequence, several web-based or nonweb- based computer software programs are publicly available for predicting miRNAs and their targets have been devised in order to predict targets for follow up experimental validation. Even though many computational methods for the identification of miRNA may have its own limitations, but there is no other option now other than to use computational methods for miRNA predictions. The next step in miRNA research is to identify and experimentally validate their mRNA targets. Since direct experimental methods for discovering miRNA targets are lacking, a large number of target prediction algorithms have been developed. Our results from this study suggests that the application of computational algorithms, PupaSuite and UTRscan analysis might provide an alternative approach to select target SNPs by understanding the effect of SNPs on the functional attributes or molecular phenotype of a protein. Our result also endorses a study with an in vivo experimental protocol. Studies using SNPs to probe the genetic basis of human disease can provide insights into susceptibility to a disease, modification of the phenotype of a monogenic disease, and response to pharmacologic treatment. The functional analysis in this study may be a good model for further research in genetically inherited disease.
We have presented computational tools for the identification of miRNA and their target mRNA in colon cancer. We tried to predict the functional roles of SNPs in mRNA region. Based on this, we derived at the following conclusions: Among the twenty genes selected for our analysis, miRNAs and their target mRNA are exhibited only by ten genes. Of these, only five genes showed multiple miRNA interactive sites for single mRNA. Out of thirty targeted SNPs by miRNAs, only seven SNPs disrupted the exonic splicing enhancers, three SNPs disrupted the exonic splicing silencers while thirteen SNPs showed no functional significance.Six SNPs exhibited functional pattern change of 15-LOX-DICE in un-translated regions. We emphasize that our approach in selecting miRNAs and their target mRNA in colon cancer using computational tools is of significant importance and the same methodology could be adapted to other types of cancer genes also. Evaluation of target mRNA functional role will be a major challenge of future studies in the field of cancer biomarker research and other types of disease.
The authors thank the management of Vellore Institute of Technology for providing the facilities to carry out this work. The authors take this opportunity to thank the reviewers for their invaluable comments and suggestions to make this manuscript more readable and meaningful.