Pancreatic Disorders & Therapy
Open Access
ISSN: 2165-7092
ISSN: 2165-7092
Research Article - (2013) Volume 3, Issue 2
Pharmacological ascorbate induces autophagy in pancreatic cancer, which is characterized by accumulation of autophagosomes and the processing of microtubule-associated protein light chain 3 (LC3) to the lipidated form, LC3- II. Prior studies have demonstrated LC3-II upregulation in pancreatic cancer cells after gemcitabine chemotherapy or ionizing radiation. The aim of this study was to determine the role of autophagy in pharmacological ascorbate-induced cytotoxicity. Pancreatic cancer cells were generated to stably overexpressLC3 fused to a green fluorescent protein (MIA PaCa-2 LC3-GFP). MIA PaCa-2 LC3-GFP cells had a higher LC3-II/LC3-I ratio at 6 and 24 hours and demonstrated a clonogenic survival advantage after pharmacological ascorbate treatment compared to parental cells. In cells with knockdown of Atg5, there was increased susceptibility to pharmacological ascorbate-induced cytotoxicity. Our data indicate autophagy may be an important survival mechanism after pharmacological ascorbate-induced cytotoxicity, while impairment of autophagy can re-sensitize cells to ascorbate. LC3 over expression has a protective effect on cell survival after ascorbate treatment. Autophagy-modulating drugs may be an important adjunct for enhancement of ascorbate-induced cytotoxicity as well as a potential therapy for re-sensitization of chemoradiation-resistant tumors.
<Keywords: Autophagy; Ascorbate; Reactive oxygen species; Microtubule-associated protein light chain 3; Pancreatic cancer
Pancreatic adenocarcinoma is one of the deadliest cancers due to delayed diagnosis and rapid metastatic progression with 5-years survival rates between 2-6%. Despite increases in technology, 5-years survival rates have remained stagnant since 1975 [1]. A better understanding of pancreatic cancer’s resistance to chemotherapy, as well mechanisms for progression and metastasis is greatly needed to make progress in treating this disease.
Autophagy may be a mode of cell death as well as a mechanism of cell survival [2-7]. Autophagy is a crucial cellular process allowing for routine turnover of cellular proteins and organelles via lysosomal degradation [4].When autophagy is triggered, a double membrane structure known as the isolation membrane forms in the cytosol. The proteins targeted for degradation are engulfed into this isolation membrane to form the autophagosome. The autophagosome then undergoes elongation, and microtubule-associated protein light chain 3 (LC3) is recruited to the autophagosome membrane. The autophagosome fuses with a lysosome to form the autolysosome and allow degradation of its contents by lysosomal enzymes [8,9]. Key steps in forming the appropriate autophagy machinery [8] include Atg12 conjugation with Atg5 which is required as an autophagosome precursor for isolation membrane formation [8]. Also required is the conversion of LC3-I to the lipidated form LC3-II, the form which localizes to the autophagosome membrane throughout the autophagy process and thus can be used as a marker to monitor autophagy [8,10]. Without either of these two steps, autophagy cannot occur properly. For instance, Kuma et al. has shown that mice require upregulation of autophagy in the immediate post-natal period. When Atg5-null mice are born, they die within the first 24 h due to their inability to upregulate autophagy as a survival mechanism during the initial starvation period prior to feeding [11].
The process of LC3-I lipidation to form LC3 -II is also essential for autophagy. LC3-II remains on the inner and outer membranes of the isolation membrane and subsequent autophagosome throughout the entire process of autophagy. LC3-II is only released after autolysosome fusion to prevent degradation and allow its recycling [8]. Sou et al. demonstrated severe impairment of LC3 lipidation as well as suppressed Atg5-Atg12 conjugation when mice were generated that lack Atg3, an enzyme responsible for LC3 conjugation. These mice had malformed autophagosomes and again died within 24 h after birth [12]. Taken together, these studies show that both Atg5 and LC3 are essential for isolation membrane formation.
The role of autophagy in cancer remains controversial. Autophagy induced by protein starvation can act as an adaptive response to nutrient deprivation [4], which could be an important role for autophagy in a rapidly growing tumor outgrowing its nutrient supply. Many cancers demonstrate dysregulation of basal autophagy levels. For example, hepatocellular carcinomas have a depressed level of autophagic protein turnover at baseline, [13,14], yet this diminished protein turnover yields less protein loss and provides a survival advantage in proteinstarved conditions [14]. In this instance, an abundance of autophagy would be detrimental to these cells, and their capability to survive relies on the ability to down regulate autophagy. In contrast, upregulation of autophagy has been demonstrated as a survival mechanism for other cell types. Pancreatic adenocarcinomas have high basal levels of autophagy [15]. In pancreatic tumors, high levels of LC3 expression have been correlated with poor clinical outcome and shorter time to tumor recurrence [16]. In radio-resistant breast cancer cells and glioma stem cells, high levels of autophagy correlate with cell survival after ionizing radiation [17,18]. Similarly, inhibition of autophagy increases apoptotic cell death in pancreatic cancer and leads to growth suppression in tumor xenografts [5,19].Thus autophagy may play a critical role in cancer cell progression and adaptation to survival in low nutrient or oxidative stress conditions.
It is clear that aberrant autophagy signaling is present in multiple cancer types and likely that cancer cells may require different levels of autophagy during different stages of oncogenic transformation and disease progression. It has been debated whether autophagy is a mechanism of cell death versus cell survival, but there has been some evidence showing autophagy as a continuum. For example, in a C. elegans model, physiologic levels of autophagy promoted optimal survival, but both insufficient and excessive levels of autophagy led to starvation hypersensitivity [20]. White has recently suggested that autophagy’s contribution to cell death versus cell survival is very context-dependent [7].
There are multiple known inducers of autophagy such as protein starvation, rapamycin, and hydrogen peroxide (H2O2). Previous studies have shown that pharmacological ascorbate, by its autoxidation to H2O2, induces autophagy in pancreatic cancer cells [21]. However, there has been conflicting evidence on the mode of cell death after ascorbate-induced cytotoxicity depending on cell type. We have previously demonstrated a depletion of intracellular ATP after 1 h ascorbate treatment, as well as an initial peak in apoptosis followed by an increasing fraction of necrotic cells at later time points [21]. Treatment with caspase inhibitors does not reverse ascorbate-induced cytotoxicity, suggesting a caspase-independent mechanism of cell death [21]. Numerous studies in multiple tumortypes also support a caspaseindependent mechanism of cell death after ascorbate treatment [22-25]. However, in our initial studies of ascorbate-induced cell death in pancreatic cancer, we also showed an increase in markers for autophagy that occurred simultaneously with caspase-independent cell death [21], which may also occur in prostate cancer cells [22].
We hypothesize that the abundant H2O2 flux generated by pharmacological doses of ascorbate, in addition to causing necrosis, may activate autophagy as a survival response to protect cells from oxidative damage.
Cell culture
Pancreatic cancer cells were purchased from American Type Culture Collection (Manassas, VA). MIA PaCa-2 cells were maintained at 37°C in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum and 1% penicillinstreptomycin. MIA PaCa-2 LC3-GFP cells were generated as previously described [21]. Briefly, MIA PaCa-2 cells were transduced to express a fusion protein of LC3 and green fluorescent protein (GFP). This fusion protein serves as a detector for autophagy. The protein distribution is spread diffusely through the cytoplasm at baseline. When cells are treated with an autophagic stimulus, the fusion protein appearance becomes more punctate, indicating LC3-I conversion to its lipidated form LC3-II, which localizes to the autophagosome membrane. Cells were selected for LC3-GFP expression by growth in 400 μg/mL hygromycin for 10 d and then maintained in DMEM supplemented with 10% fetal bovine serum and 200 μg/mL hygromycin. GFP positivity was confirmed by flow cytometry for green fluorescence and with propidium iodide (1 μg/mL) staining for viability. Immediately prior to our planned experiments with these cells, the viable cell population was found to be 91% GFP positive.
Western blot
Protein concentration from cell lysates was quantified by Bio Rad DC™ colorimetric-based protein assay (Life Science, Hercules, CA) according to the manufacturer’s instructions. The protein was loaded in equal quantities and electrophoresed in a 4-20% SDS-polyacrylamide gel followed by electro-transfer to an Immobilon PVDF membrane (EMD Millipore, Billerica, MA). Non-specific protein binding was blocked using 5% nonfat powdered milk in PBS-tween. Blots were treated overnight at 4°C with primary antibodiesto LC3 (1:1000; MBL International, Woburn, MA), Atg5 (1:000; Abcam, Cambridge, MA), or actin (1:4000; Sigma-Aldrich, St. Louis, MO) as a loading control. Anti-mouse IgG or anti-rabbit IgG conjugated to peroxidase was used as a secondary antibody (1:25,000; Sigma-Aldrich, St. Louis, MO).The washed blot was treated with Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Rockford, IL) and exposed to radiographic film.
Clonogenic survival
L-ascorbic acid was purchased from Macron Chemicals (Center Valley, PA). Stock solutions of ascorbate (1.0 M, pH7.0) were made under argon as previously described [26]. For all ascorbate treatments, cells were placed in fresh media and treated with ascorbate for 1 h at 37°C. Media was changed at 1 h, and cells were harvested at the appropriate time points for Western blot or clonogenic survival assay. For groups treated with rapamycin, cells were pre-treated with 5 μM rapamycin (Sigma-Aldrich, St. Louis, MO) for 24 h prior to ascorbate treatment. Immediately after treatments, cells were washed in HBSS, trypsinized, and counted. Cells were plated in 6-well plates with 300 cells/well in 4 mL complete medium and incubated at 37°C for 10 d. Colonies were fixed in 70% ethanol and stained with Coomassie blue. Those colonies containing greater than 50 cells were scored, and surviving fraction was determined.
Confocal microscopy
Cells were prepared for confocal microscopy as described previously [21]. Briefly, cells were grown on a coverslip and treated with ascorbate (2 mM) with or without pre-treatment with rapamycin (5 μM). Cells were fixed and stained. TO-PRO-3 (1:2000) was used as a nuclear counterstain, and the cells’ inherent GFP expression was used as a marker for LC3 localization. The samples were imaged with a confocal microscope equipped with a Kr/Ar laser as described previously [21].
Flow cytometry
Cells were treated with ascorbate (2 mM) with or without rapamycin pre-treatment (5 μM) and prepared for flow cytometry as described previously [21]. Briefly, cells were resuspended in propidium iodide (1 μg/mL) and run on a BD Calibur flow cytometer with gating for viability (PI negative) and GFP positivity. Mean fluorescence intensity (MFI) of GFP positive cells were quantified.
Atg5 knockdown
To examine the effects of ascorbate-induced cytotoxicity during inhibition of autophagy, we obtained TEM 4-18 prostate cancer cells [27] and TEM 4-18 cells treated with a short hairpin construct against Atg5 (TEM 4-18shAtg5) from Dr. Michael Henry (University of Iowa). Prostate cancer cell lines were maintained in DMEM/F12 (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 200 μg/mL hygromycin, and 1 μg/mL puromycin.
Effect of pharmacological ascorbate in pancreatic cancer cells that over express LC3. First, MIA PaCa-2 and MIA PaCa-2 LC3-GFP cells were characterized for basal LC3 expression via Western blot (Figure 1A). We observed MIA PaCa-2 LC3-GFP cells not only express this fusion protein but overexpress LC3 in large quantities compared to parent MIA PaCa-2 cells. Next, MIA PaCa-2 and MIA PaCa-2 LC3- GFP cells were treated with ascorbate (2 mM) and harvested for Western blot at various time points (0-24 h) (Figure 1B). Western blot demonstrated an increase in the LC3-II/LC3-I ratio at 6 h and 24 h when compared to 0 h in the MIA PaCa-2 LC3-GFP cells. This was quantified by densitometry (Figure 1C). MIA PaCa-2 LC3-GFP cells expressed 20% higherLC3-II/LC3-I ratio at 24 h after ascorbate treatment relative to their baseline compared to parental MIA-PaCa-2 cells, which maintained a steady LC3-II/LC3-I ratio over time. This indicates that over expression of LC3 protein may potentially allow for a higher autophagy capacity.
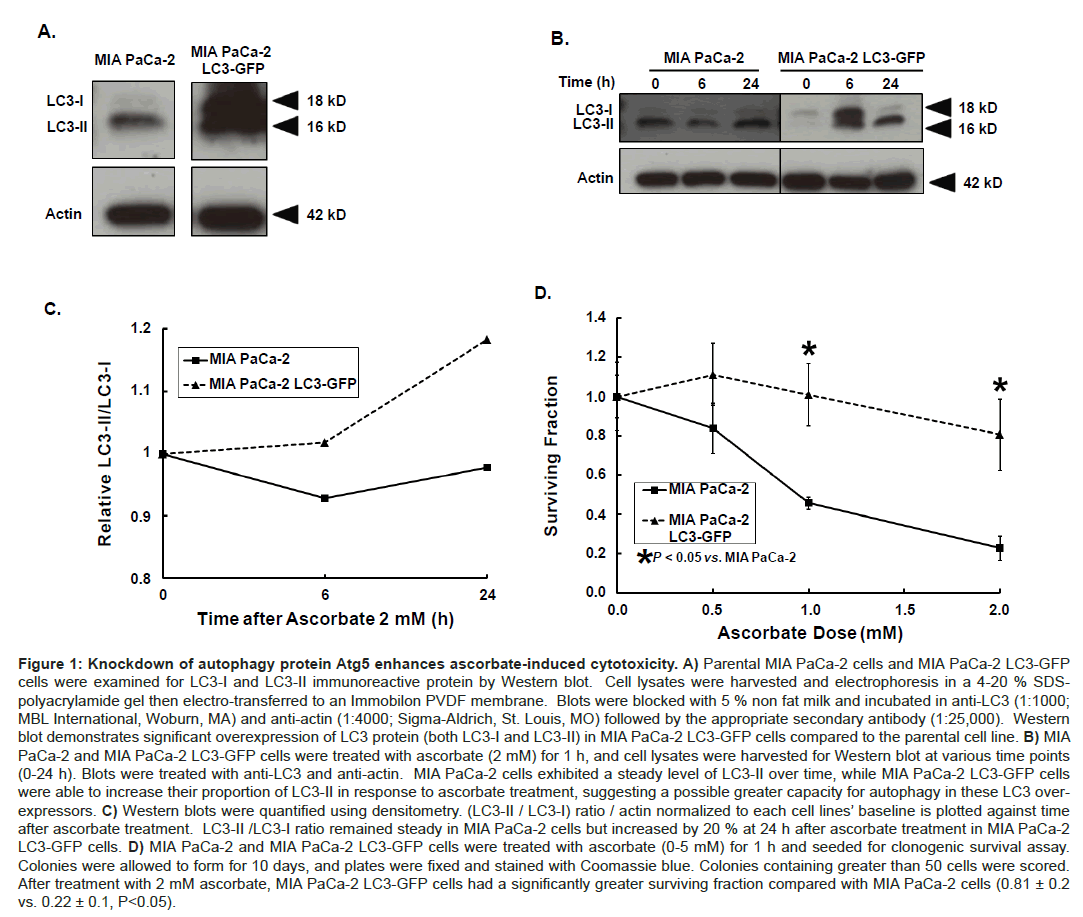
Figure 1: Knockdown of autophagy protein Atg5 enhances ascorbate-induced cytotoxicity. A) Parental MIA PaCa-2 cells and MIA PaCa-2 LC3-GFP cells were examined for LC3-I and LC3-II immunoreactive protein by Western blot. Cell lysates were harvested and electrophoresis in a 4-20 % SDSpolyacrylamide gel then electro-transferred to an Immobilon PVDF membrane. Blots were blocked with 5 % non fat milk and incubated in anti-LC3 (1:1000; MBL International, Woburn, MA) and anti-actin (1:4000; Sigma-Aldrich, St. Louis, MO) followed by the appropriate secondary antibody (1:25,000). Western blot demonstrates significant overexpression of LC3 protein (both LC3-I and LC3-II) in MIA PaCa-2 LC3-GFP cells compared to the parental cell line. B) MIA PaCa-2 and MIA PaCa-2 LC3-GFP cells were treated with ascorbate (2 mM) for 1 h, and cell lysates were harvested for Western blot at various time points (0-24 h). Blots were treated with anti-LC3 and anti-actin. MIA PaCa-2 cells exhibited a steady level of LC3-II over time, while MIA PaCa-2 LC3-GFP cells were able to increase their proportion of LC3-II in response to ascorbate treatment, suggesting a possible greater capacity for autophagy in these LC3 overexpressors. C) Western blots were quantified using densitometry. (LC3-II / LC3-I) ratio / actin normalized to each cell lines’ baseline is plotted against time after ascorbate treatment. LC3-II /LC3-I ratio remained steady in MIA PaCa-2 cells but increased by 20 % at 24 h after ascorbate treatment in MIA PaCa-2 LC3-GFP cells. D) MIA PaCa-2 and MIA PaCa-2 LC3-GFP cells were treated with ascorbate (0-5 mM) for 1 h and seeded for clonogenic survival assay. Colonies were allowed to form for 10 days, and plates were fixed and stained with Coomassie blue. Colonies containing greater than 50 cells were scored. After treatment with 2 mM ascorbate, MIA PaCa-2 LC3-GFP cells had a significantly greater surviving fraction compared with MIA PaCa-2 cells (0.81 ± 0.2 vs. 0.22 ± 0.1, P<0.05).
We hypothesized this increase in LC3-I processing to LC3-II may have a protective effect on cell survival if indeed autophagy allows cells to increase damaged protein turnover when faced with oxidative stress. To assess cell survival, MIA PaCa-2 and MIA PaCa-2 LC3-GFP cells were treated with ascorbate (0, 0.5, 1, and 2 mM) and plated for clonogenic survival assay. MIA PaCa-2 LC3-GFP cells were resistant to ascorbate treatment when compared to parental MIA PaCa-2 cells (Figure 1D). For example, after treatment with 2 mM ascorbate the number of colonies formed by MIA PaCa-2 cells was only 22% of the amount formed by untreated cells, whereas the number of colonies formed by MIA PaCa-2 LC3-GFP cells remained at 81% of their baseline (P<0.05). These data suggest that LC3 over expression confers resistance to ascorbate-induced cytotoxicity, and this resistance may be due to the cell’s capacity to upregulate autophagy.
Rapamycin enhances ascorbate-induced increases in autophagy in LC3 over expressing cells. Another method of evaluating LC3-I processing to LC3-II as a marker for autophagy is following the appearance of punctae in cells expressing LC3-GFP protein. As LC3-I is lipidated to form LC3-II, the fluorescence changes from a diffuse green in the cytoplasm to punctate green dots along the autophagosome membranes [10]. MIA PaCa-2 LC3-GFP cells were treated with ascorbate (2 mM) with and without rapamycin pre-treatment (5 μM). A subset of cells was prepared using TO-PRO-3 (1:2000) as a nuclear counter stain and imaged with a confocal microscope equipped with a Kr/Ar laser as described previously [21]. Confocal microscopy demonstrated increased formation of green fluorescent punctae with either ascorbate or rapamycin treatment and an even greater fluorescence with the combination treatment (Figure 2A). In another group, cells were treated with ascorbate (2 mM) with and without rapamycin pre-treatment (5 μM) and resuspended in propidium iodide (1 μg/mL) to sort for cell viability. Viable cells were examined for green fluorescence indicating GFP positivity, and mean fluorescence intensity (MFI) of the GFP positive cells was quantified as described previously [21]. Cells treated with either rapamycin or ascorbate alone had significantly greater MFI compared to untreated MIA-PaCa-2 LC3-GFP controls with an MFI of 129 ± 1. Rapamycin treatment increased MFI to146 ± 3 (P<0.05), while ascorbate treatment increased the MFI to 177 ± 4 (P<0.05). The combination treatment had an even greater increase in MFI to 195 ± 2, which was significantly higher when compared to control or treatment with either rapamycin or ascorbate alone (P<0.05 vs. control, ascorbate, or rapamycin) (Figure 2B). Taken together, these experiments suggest that induction of autophagy with rapamycin [10], enhances ascorbateinduced autophagy.
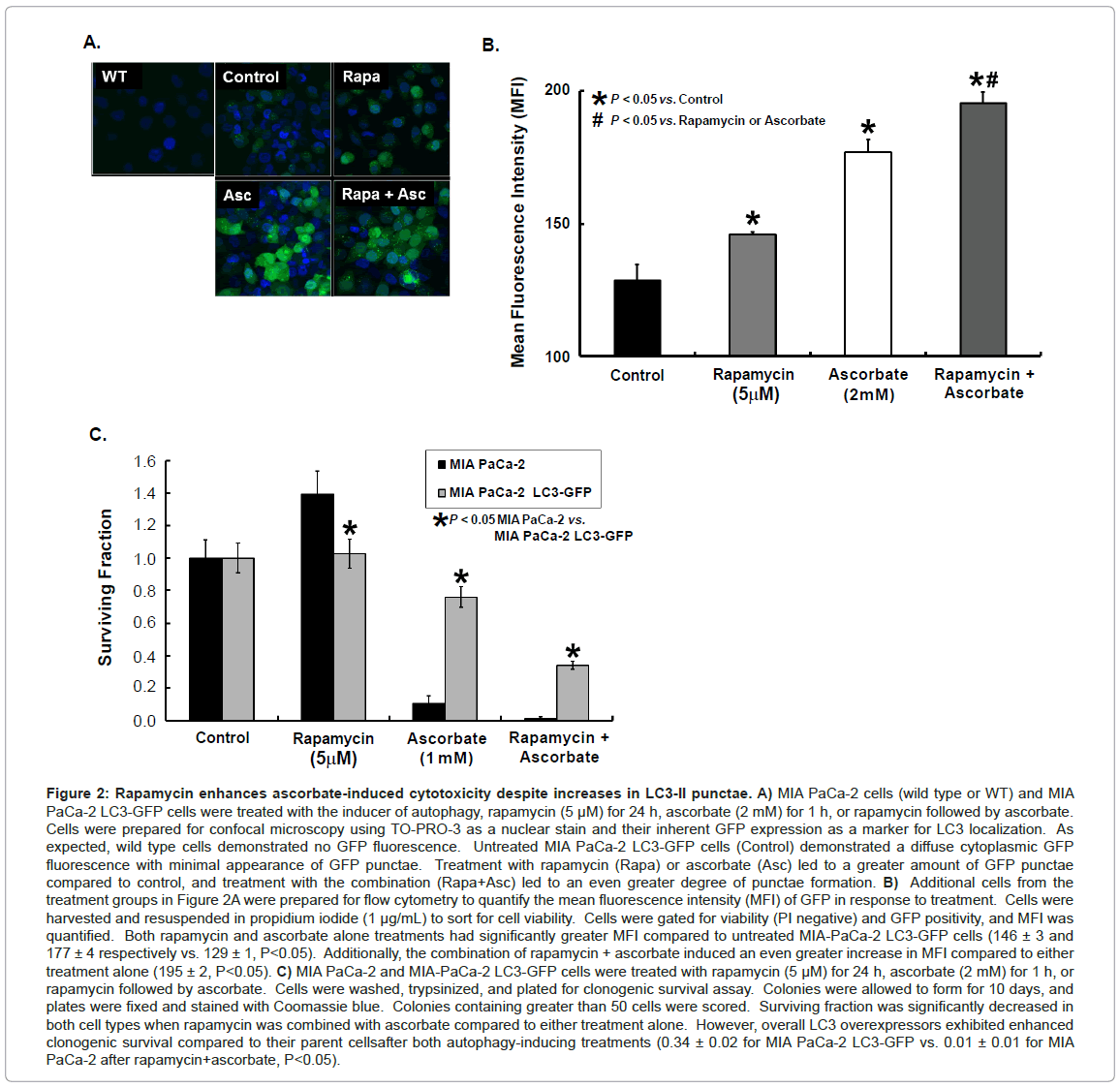
Figure 2: Rapamycin enhances ascorbate-induced cytotoxicity despite increases in LC3-II punctae. A) MIA PaCa-2 cells (wild type or WT) and MIA PaCa-2 LC3-GFP cells were treated with the inducer of autophagy, rapamycin (5 μM) for 24 h, ascorbate (2 mM) for 1 h, or rapamycin followed by ascorbate. Cells were prepared for confocal microscopy using TO-PRO-3 as a nuclear stain and their inherent GFP expression as a marker for LC3 localization. As expected, wild type cells demonstrated no GFP fluorescence. Untreated MIA PaCa-2 LC3-GFP cells (Control) demonstrated a diffuse cytoplasmic GFP fluorescence with minimal appearance of GFP punctae. Treatment with rapamycin (Rapa) or ascorbate (Asc) led to a greater amount of GFP punctae compared to control, and treatment with the combination (Rapa+Asc) led to an even greater degree of punctae formation. B) Additional cells from the treatment groups in Figure 2A were prepared for flow cytometry to quantify the mean fluorescence intensity (MFI) of GFP in response to treatment. Cells were harvested and resuspended in propidium iodide (1 μg/mL) to sort for cell viability. Cells were gated for viability (PI negative) and GFP positivity, and MFI was quantified. Both rapamycin and ascorbate alone treatments had significantly greater MFI compared to untreated MIA-PaCa-2 LC3-GFP cells (146 ± 3 and 177 ± 4 respectively vs. 129 ± 1, P<0.05). Additionally, the combination of rapamycin + ascorbate induced an even greater increase in MFI compared to either treatment alone (195 ± 2, P<0.05). C) MIA PaCa-2 and MIA-PaCa-2 LC3-GFP cells were treated with rapamycin (5 μM) for 24 h, ascorbate (2 mM) for 1 h, or rapamycin followed by ascorbate. Cells were washed, trypsinized, and plated for clonogenic survival assay. Colonies were allowed to form for 10 days, and plates were fixed and stained with Coomassie blue. Colonies containing greater than 50 cells were scored. Surviving fraction was significantly decreased in both cell types when rapamycin was combined with ascorbate compared to either treatment alone. However, overall LC3 overexpressors exhibited enhanced clonogenic survival compared to their parent cellsafter both autophagy-inducing treatments (0.34 ± 0.02 for MIA PaCa-2 LC3-GFP vs. 0.01 ± 0.01 for MIA PaCa-2 after rapamycin+ascorbate, P<0.05).
Since autophagy may protect against ascorbate-induced cytotoxicity and rapamycin enhances ascorbate-induced autophagy we next investigated the effect of rapamycin on ascorbate-induced cytotoxicity using the clonogenic survival assay. MIA PaCa-2 and MIA PaCa-2 LC3-GFP cells were treated with ascorbate (1 mM) with and without rapamycin (5 μM) pre-treatment and seeded for clonogenic survival. Treatment with ascorbate or the combination of rapamycin and ascorbate inhibited clonogenic survival compared to controls in both the parental and the LC3 over expressing cells. Additionally, LC3 over expression led to increased resistance to ascorbate-induced cytotoxicity (Figure 2C). This data suggests that LC3 over expression possibly resulting in altered levels of autophagy may serve as a protective mechanism against ascorbate-induced cytotoxicity.
Loss of Atg 5 alters ascorbate’s effects on LC3-II accumulation and clonogenic survival. Atg5-Atg12 complex is a critical component in formation of the isolation membrane or pre-autophagosome, which is necessary for the conversion of LC3-I to LC3-II and subsequent formation of the autophagosomes [8]. To examine the effects of Atg5 knockdown on autophagy, we evaluated basal autophagy levels by Western blot for LC3-I and LC3-II expression, as well as the current degree of knockdown of the Atg5-Atg12 complex (Figure 3A). To examine the effects of ascorbate on autophagy in these cells, the cells were treated with ascorbate (2 mM), and cell lysates were harvested for Western blot. At baseline, TEM 4-18shAtg5 cells had a lower amount of immunoreactive protein for Atg5-Atg12 complex and concordantly had a lower LC3-II/LC3-I ratio when compared to the parental cell line (Figure 3A). After treatment with ascorbate, both cell lines had reduced immunoreactive protein for Atg5-Atg12 complex. However, the Atg5 knockdown cells expressed a higher LC3-II/LC3-I ratio after ascorbate treatment (Figure 3A). This suggests that these cells may be capable of an Atg5-independent form of autophagy, LC3 lipidation may be able to occur through an alternative mechanism, or that ascorbate may be impairing autophagic flux rather than increasing steady state autophagy.

Figure 3: LC3 over expression leads to increased LC3-II/LC3-I ratio following ascorbate treatment and enhanced resistance to ascorbateinduced cytotoxicity. A) TEM 4-18 and TEM 4-18shAtg5 cells were used to examine the effect of ascorbate on levels of Atg5 and LC3. At baseline TEM 4-18shAtg5 cells had low levels of Atg5-Atg12 complex as well as a low LC3-II / LC3-I ratio compared to TEM 4-18 cells. However, after ascorbate treatment (2 mM for 1 h), TEM 4-18shAtg5 cells had a greater increase in LC3-II compared to the parental cells despite having undetectable levels of the Atg5-Atg12 complex. B) The cell line TEM 4-18, with increased amounts of Atg5-Atg12 complex at baseline, was resistant to pharmacological ascorbate treatment. However, the TEM 4-18shAtg5 cells showed a significant decrease in surviving fraction (65% of control, P<0.05) after ascorbate treatment, suggesting Atg-5 may be necessary for successful autophagy-induced resistance to pharmacological ascorbate cytotoxicity.
Since autophagy or at least high amounts of LC3-II were protective against ascorbate-induced cytotoxicity, we hypothesized that if cells were indeed undergoing autophagy or at a minimum LC3 lipidation in an Atg5-independent manner, both the TEM 4-18 and the TEM 4-18shAtg5 cells should have similar sensitivity to pharmacological ascorbate. However, if depressed levels of Atg5 are indicating lack of autophagy and the presence of LC3 lipidation is less significant, we would expect to see an increased cell killing in cells with lower levels of Atg5. To test this hypothesis, TEM 4-18 and TEM 4-18shAtg5 cells were treated with ascorbate (5 mM) for 1 h and seeded for clonogenic survival assay (Figure 3B). Surviving fraction for TEM 4-18 cells after treatment with ascorbate (5 mM) was not significantly different from baseline. However, the surviving fraction of TEM 4-18shAtg5 cells after ascorbate treatment was decreased to 65% of baseline (Figure 3B), suggesting that Atg5 is important in the mechanism for survival after ascorbate treatment. Despite the presence of processing of LC3-I to LC3-II, sufficient autophagy for survival may not be occurring in the absence of Atg5.
We have shown that over expression of LC3 leads to an increased LC3-II/LC3-I ratio yielding an increased cell survival in pancreatic cancer cells. This suggests that autophagy may be a survival mechanism in pancreatic cancer. There has been conflicting evidence on whether autophagy is a cell death mechanism or a cell survival mechanism in cancer. Chen et al. demonstrated autophagic cell death as a mechanism of ascorbate-induced cytotoxicity and recently has shown that knockdown of Bif1, which is a positive mediator of autophagy, rescues ascorbate-induced cell death. These findings imply suppression of autophagy inhibits ascorbate-induced cell death [22]. However, autophagy may also be a mechanism for tumor cell survival and resistance to therapy. Two commonly used therapies in pancreatic cancer include ionizing radiation and gemcitabine chemotherapy. Chaachouay et al. revealed autophagy as a mechanism for resistance to ionizing radiation in breast cancer by demonstrating increased LC3-II/ LC3-I ratio in radio-resistant breast cancer cells, as well as by preventing radio-resistance with the inhibitors of autophagy,3-methyladenine (3-MA) and chloroquine [17]. In addition, Mukubou et al. displayed a similar up regulation of LC3-II in pancreatic cancer after treatment with gemcitabine and ionizing radiation [28]. Work by Papademetrio et al. also supports autophagy as a mechanism responsible for pancreatic cancer’s refractory response to gemcitabine [29]. Furthermore, Li et al. recently showed that Beclin-1 silencing upregulates autophagy in pancreatic cancer leading to decreased apoptosis after gemcitabine treatment; this again supports that autophagy may play a role in tumor cell survival when pancreatic cancers are refractory to treatment [30]. Our work also suggests a protective role for autophagy during pharmacological ascorbate treatment. We have shown impairment of autophagy through Atg5 knockdown enhances prostate cancer cell killing by pharmacological ascorbate.
Nonetheless, our current studies with the autophagy inducer rapamycin demonstrate a differing role for autophagy as an effector in cell death. Rapamycin enhanced ascorbate-induced increases in autophagy as seen by increases in LC3-GFP punctae. However, the combined treatment also led to increased cytotoxicity with ascorbate. Regulation of autophagy is a complex process, which is not yet fully understood, but our dissonance of results concerning autophagy as well as an abundance of literature describing different cellular consequences of autophagy depending on different contexts [5,7,20,22,30] suggests a model where perhaps smaller amounts of autophagy can be protective. However, when the cell autophagy machinery is overwhelmed, cell death can occur.
LC-3 over expression conferred a survival advantage in MIA PaCa- 2 cells when exposed to ascorbate. However, the ability to process LC3-I to LC3-II alone was insufficient to prevent ascorbate-induced cytotoxicity in prostate cancer cells with Atg5 knockdown. This poses a number of questions. First, is this an issue of autophagic flux versus autophagy steady state levels; secondly, how reliable is LC3 as a marker of autophagy; and thirdly, does LC3 have an additional function in cell survival aside from its role in autophagy? Interestingly, it has previously been shown that some LC3-II can be generated via an autophagyindependent mechanism in the setting of mutants (such as Atg5-null cells) or of pharmacological inhibition of autophagy [31]. For example, LC3-II has been detectable even in the presence of RNA-interference of other Atg proteins such as Atg13 and Atg14 [32,33], even though these proteins are thought to be required for complete autophagy to occur. Additionally, the accumulation of LC3-II occurs despite the presence of 3-MA inhibition of autophagy, suggesting LC3-II alone may be necessary but not sufficient for autophagy to occur [34]. Other reports have demonstrated an Atg5-independent form of macroautophagy in mouse cells lacking Atg5 or Atg7. However, in this instance lipidation of LC3 and accumulation of LC3-II was not seen as in our present study [35].
Others have argued whether LC3-II should be used as a marker for autophagy.3-MA, despite its blockage of the autophagy process, traps LC3-II in its active form [34], suggesting that LC3-II may not be a reliable indicator of autophagy. LC3 may also have functions outside of the autophagy pathway, as LC3-GFP can interact directly with p62 at protein aggregates apart from the autophagosomes [36].
This conflicting information on the presence of LC3-II raises the question of what is actually occurring with LC3 over expression. Is LC3- II indicative of increased autophagy or is the LC3-II itself participating in some other cellular process? Clinically, high levels of LC3 expression correlate with aggressive tumor biology, metastasis, and poor prognosis when present in melanoma tumor specimens [37]. Similarly, in a mouse model of sepsis LC3-GFP over expression improves survival due to an enhanced clearance of autophagosomes [38], which could be one possible mechanism to explain enhanced survival after ascorbate treatment in our LC3-overexpressing cells.
Our current study demonstrates that higher levels of autophagy induced by rapamycin and ascorbate result in cell death. Paradoxically, LC3 over expression results in protection from cell death. Similarly, blockage of Atg5 results in sensitization to pharmacological ascorbateinduced cytotoxicity. Taken together, our work suggests ascorbate and its generation of H2O2 could be acting at multiple levels on the autophagy pathway, which includes multiple kinases that could be potential targets for 2-electron redox reactions with H2O2. LC3 over expression results in a protection against ascorbate-induced cytotoxicity, but we have not fully elucidated whether this is secondary to increased autophagy or an autophagy-independent function of LC3. Autophagy remains a complicated cellular process with the mechanism not yet fully understood, but with further study autophagy-modulating drugs may become an adjunct for enhancement of ascorbate-induced cytotoxicity as well as an important therapy for re-sensitization of chemo-radiationresistant tumors.
We thank Dr. Michael Henry and Jones Nauseef for their generous gift of TEM 4-18 and TEM 4-18shATG cells. We acknowledge the University of Iowa Central Microscopy Research Facility for assistance with confocal microscopy. We thank Brett Wagner for preparing the ascorbate. Supported by NIH grants U01166800, and T32 CA148062, and the Medical Research Service, Department of Veterans Affairs 1I01BX001318-01A2.