Journal of Hepatology and Gastrointestinal disorders
Open Access
ISSN: 2475-3181
ISSN: 2475-3181
Research Article - (2022)Volume 8, Issue 5
Objective(s): Hirschsprung disease (HSCR) is a developmental disorder characterized by the absence of ganglion cells in the gastrointestinal tract, resulting in intestinal obstruction. HSCR has two significant forms: sporadic and familial/syndromic. This study aimed to investigate the genetic basis of HSCR in an Iranian extended family to explore the causal mutations.
Materials and Methods: DNA extraction was carried out from blood specimens of the twelve affected and unaffected family members. Then, Whole Exome Sequencing (WES) was performed for patients IV-V, and the identified variant was validated by Sanger sequencing in the proband IV-V. Finally, a segregation analysis was conducted on the parents and other affected and unaffected family members.
Results: We have identified a novel heterozygous nonsense mutation with the standard filtering protocol, p.Y314X, at exon 5 of the RET gene. This devastating variant was manifested in the proband with long-segment form and other phenotypes such as single kidney, absence of peritoneum, and the face’s pigmentation. However, this variant has been segregated into the mother, the grandmother, and the aunt. These members of the family did not show HSCR. Furthermore, this variant was detected in other family members (II-IV and III-XII) with chronic constipation and no HSCR.
Conclusion: The present study has found a novel nonsense variant at the RET gene associated with a wide range of phenotypes and incomplete penetrance. The synergistic effects of rare and common variants among known and unknown disease susceptibility genes lead to variable severity. Such information is vital for proper genetic counseling in familial HSCR.
Hirschsprung disease; Whole-Exome sequencing; RET novel mutation; Extended pedigree; Incomplete penetrance
HSCR: Hirschsprung disease; ENS: Enteric Nervous System; MAF: Minor Allele Frequency; S-HSCR: Short-Segment Hirschsprung disease; L-HSCR: Long-Segment Hirschsprung disease; TCA: Total Colonic Aganglionos; CDS: Coding Sequence; PND: Prenatal Diagnosis; WES: Whole-Exome Sequencing; Indels: Short insertions and deletions; SNVs: Single Nucleotide Variants; LOF: Loss-of-Function; ENCC: Enteric Neural Crestderived Cell; NMD: Nonsense-Mediated mRNA Decay; RTKs: Receptor Tyrosine Kinase; VUS: Variant Uncertain Significance; CAKUT: Congenital Anomalies of the Kidney or Urinary Tract
Hirschsprung’s disease (HSCR) is recognized as an intestinal aganglionosis and an uncommon congenital anomaly caused by the full absence of intestinal Enteric Neural Crest-Derived Cell (ENCC) progenitors during the development of the fetal intestine [1]. The global prevalence of this disease is approximately 1 per 5000 live births, while it significantly varies among different ethnic groups [2]. HSCR has a strong genetic basis, and it is reported that different forms of the disease have a specific pattern of inheritance [3].
There are three main types of HSCR based on the size of the aganglionic segment, known short-segment (S-HSCR; 80% of the cases), long-segment (L-HSCR; 15% of the cases), and Total Colonic Aganglionosis (TCA; 5% of the cases). S-HSCR, the most common type of disease, is highly heritable (>80%) and shows variable expression with a complex form of inheritance and low sex-dependent penetrance [4,5]. Nevertheless, the other types of disease appear with a familial/syndromic basis (30% of the cases) along with incomplete penetrance (autosomal inheritance) [6].
Genetic studies of HSCR have identified extensive genetic heterogeneity. Currently, more than 17 susceptibility genes, including RET, GDNF, NRTN, and SOX10 have been known to be related to HSCR [7]. These genes encode for protein components mediated in particular signaling pathways that are involved in developing the Enteric Nervous System (ENS). The RET protooncogene is a principal gene with >80% of all known mutations related to HSCR and is responsible for nearly 50% of familial and up to 15% of sporadic cases. However, the other genes may contribute to approximately 10% of the HSCR cases [8].
RET encodes a receptor tyrosine kinase that regulates the proliferation, differentiation, and migration of the enteric neural crest cells to ENS throughout the length of the fetal intestine [9]. In HSCR patients, more than 100 RET mutations have been reported. These mutations, which are scattered throughout the entire Coding Sequence (CDS), are divided into two groups. These groups consist of nonsense and frameshift mutations as the first class that can encode a truncated protein, and the missense mutations and small in-frame deletions as the second one, which can lead to nonfunctional protein production. The responsible mechanism in HSCR cases is mainly haploinsufficiency/loss of function mutation [10]. However, gain-of-function mutations of the RET gene are the most common causes in patients suffering from hereditary thyroid cancers [11]. Additionally, a common variant of rs2435357 located in a gut-specific RET enhancer element in intron 1 can disrupt the binding of SOX10 and acts synergistically on HSCR disease risk [12].
Although HSCR is usually diagnosed early after birth [13], it is observed that treatment failure of this disease at this age is often fatal. Therefore, determining the functional consequences of these mutations and understanding genotype-phenotype correlation can help the better management of disease and be used in Prenatal Diagnosis (PND), especially in the case of familial HSCR. The present study aimed to uncover the novel pathogenic variant of HSCR in an Iranian extended family with various manifestations such as inherited chronic constipation and congenital malformations using Whole-Exome Sequencing (WES) as a high-throughput and cost-effective method. To the best of our knowledge, this is the first study investigating pathological mutations of susceptible genes in Iranian HSCR patients.
Families’ enrolment and sample preparation
Seven affected and five unaffected family members have been recruited and blood sample were taken. The informed consent form has been signed by all the subjects. The diagnosis of HSCR was confirmed by an experienced gastroenterologist based on their symptoms after a clinical examination. DNA was extracted from the peripheral blood using the Salting out procedure [14]. This study is approved by the Research Ethics Committee of the Iran University of Medical Sciences.
Raw data analysis and variant calling
Exome capture and library preparation were performed for patients IV-V, using Agilent SureSelect V6 Target Enrichment Kit (Agilent, Santa Clara, CA, USA). Whole Exome Sequencing (WES) of pairedend 150 base-pair (bp) reads was carried out using IlluminaHiSeq 4000 platform. Briefly, the reads were aligned to the human reference genome (GRCh37/hg19 assembly) using BWA software version 0.7.12 [15]. Next, the duplicate and low-quality reads were removed, and Single Nucleotide Variants (SNVs) and short insertion–deletions (Indels) were called using the GATK software package to apply base quality score recalibration (QBase<20).
VCF data were annotated utilizing the wannovar tool. Our workflow of variant filtering is shown in Figure 1. In the beginning, the annotated file was prioritized by an in-house pipeline with zygosity of variants (homozygous and heterozygous). Then, common variants with the minor allele frequency of >0.05 in dbSNP137, 1000 Genomes database, NHLBI Exome Sequencing Project, ExAC database, and genomAD database were filtered out. The prediction of the potential impact of rare variants of coding-sequence on the function or structure of the protein was made according to six insilico bioinformatics tools, including SIFT, Mutation Taster [16,17], CADD score, DANN score and PolyPhen2 [18-20]. In addition, conservation studies were conducted using the GERP score [21]. Among the filtered variants, nonsense, splice site, frameshift, inframeshift, and missense mutations of the susceptible genes for HSCR were selected and checked in HGMD and ClinVar with the help of PubMed and OMIM databases for ranking the genes related to HSCR. To find variants related to the patient phenotype, we used HPO terms extracted from the HPO database; such as Aganglionic Megacolon/Hirschsprung Disease: HP:0002251. The identified variants were classified and interpreted based on the ACMG-AMP 2015 standards and guidelines facilitated by the varsome tool [22,23].

Figure 1: The workflow of variant filtering. The variants’ annotation and the filtering steps were done based on Minor Allele Frequency (MAF), the mode of inheritance, and the pathogenicity scores by the prediction tools. The candidate variants were confirmed by Sanger sequencing.
Pathogenic mutations validation
The identified potential pathogenic variants were validated by Sanger sequencing in the proband IV-V, and segregation analysis was performed on the parents and other affected and unaffected family members. For this purpose, primers of the specific exons were designed using Primer3, and PCR products were sequenced by ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Chromas v2.33 software was used to monitor sequencing results (Technelysium, Tewantin, Qld, Australia).
Clinical features of patients
The proband IV-V was a 10-year-old girl who suffered from Hirschsprung disease and congenital right kidney aplasia. Her birth measurements (weight: 3000 g and length: 48 cm) and the neonatal history were standard. However, after six months, she manifested the symptoms of abdominal distension, vomiting, diarrhea, fever, and chronic constipation that lead to hospitalization. Histological examination of the proband showed aganglionic myenteric plexus, rectal, sigmoid, splenic flexure, and considerable intestinal and appendix involvement diagnosed as HSCR. Then, she underwent a colectomy and was given antibiotic drugs. More investigation into family history revealed that the proband’s mother (III-IV) has a single kidney (renal agenesis). The subjects II-III, II-V, II-VII, III-VIII, and III-X died after birth with congenital malformations such as closed anus and intestine obstruction. Furthermore, other family members (I-II, II-IV, II-VI, III-V, III-XII) had been suffering from severe chronic constipation throughout their lives. The family pedigree and other abnormalities along with HSCR in family members is shown in Figure 2.

Figure 2: The family pedigree with HSCR and other abnormalities along in family members. Note: ( ) HSCR, (
) HSCR, ( ) Single Kidney, (
) Single Kidney, ( )
Constipation, (
)
Constipation, ( ) Diarrhea, (
) Diarrhea, ( ) Pigmentation of the face, (
) Pigmentation of the face, ( ) Premature ovarian failure, (
) Premature ovarian failure, (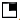 ) Closed anal end, (
) Closed anal end, (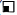 ) Absent of peritineum
) Absent of peritineum
Rare variants in splicing or coding regions
Whole Exome Sequencing (WES) was performed to find the genetic variants of IV-V proband related to HSCR. A 7.2 Gigabase sequence was generated, and the minimum mean coverage of target regions consists of exons, and the first and last 20 bps of introns were 97.7%, 96.2%, and 92.2% covered at >1X, >5X, and >10X, respectively. 113,077 variants were detected in the IV-V proband’s VCF file, which includes 103,620 substitutions and 9,200 indels (insertion/deletion) variations in the exome analysis. As explained before, we performed the filtering of variants based on frequent variants found in different populations, zygosity (inheritance pattern of HSCR could be autosomal dominant or recessive), and pathogenic impacts of variants using bioinformatics tools such as CADD-PHRED score and mutation taster. Firstly, 53,063 homozygous and 59,839 heterozygous variants were detected after zygosity filtering. To identify the rare variants, we gained MAFs from population databases (MAF<0.05).
Classification/annotation of novel variations
Several in-silico analyses were performed to evaluate the pathogenicity of dominant and recessive variants. The variants with CADD-PHRED score >10, disease-causing variants evaluated by Mutation Taster, pathogenic alterations with DANN Score value range of 0 to 1, and variants with GERP score ranging from -12.3 to 6.17 obtained from conservation studies were selected. Finally, a novel heterozygous stop-gain variant p.Y314X or c.C942A in exon 5 of the RET gene was identified in the proband IV-V (NM_020975, chr10-43601898).
Bioinformatics prediction analysis by vaesome and the manual examination of the ACMG-AMP 2015 guidelines have considered this variant as pathogenic as follows:
• A single base exchange results in a premature termination codon, acting as a null variant leading to Nonsense-Mediated mRNA Decay (NMD) pathway or a truncated protein, which by itself robust the evidence in favor of pathogenicity (PVS1).
• This variant has been removed from control variant lists of Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium, and GenomAD databases (PM2 criteria).
• Multiple lines of computational evidence support the deleterious effect of this variant on the gene or gene product (conservation, evolutionary, splicing impact, etc.) (PP3). The results of in-silico computational analysis and frequency of the identified variant in population databases are presented in Table 1. We validated this variant by Sanger sequencing on proband IV-V followed by cosegregation analysis in all available family members.
| Variant Definition | In-Silico Predictive Tool | Population Database | |||
|---|---|---|---|---|---|
| Variant name | c.942C>A | Variant name | c.942C>A | Variant name | c.942C>A |
| Protein Change | p.Y314X | CADD (PhredScore) | 36 (Deleterious) | 1000 GP | - |
| Variant Location | Exon 5 | DANN | 0.99 | ExAC | - |
| Chromosome Position (GRCh37) | Chr10:43601898 | GERP | 5.3 | GenomAD | - |
| Zygosity | Heterozygote | Mutation Taster | Disease-Causing | ESP | - |
Other rare variants in hirschsprung disease-associated genes
Several rare variants with MAF<0.05 were identified in the patient (IV-V), which are related to other HSCR-associated genes (literature reviews and original articles at PubMed, OMIM, and phenolyzer databases were searched for ranking the genes associated with HSCR). Table 2 shows the results of in-silico analysis, allele frequency obtained from GenomAD, and results of ACMG prediction analysis using varsome. Additionally, particular heterozygous variants with the minor allele frequency of <0.05 in GenomAD databases, and CADD score >10 were detected after filtering values. The potential impact of these variants on the function or structure of the protein and conservation studies were evaluated using bioinformatics tools, including SIFT (tolerated/damaging), mutation taster (polymorphism/disease-causing), GERP (ranging from -12.3 to 6.17, with 6.17 for the most conserved variants), and DANN (the value range is 0 to 1, with 1 for the variants predicted to be the most damaging). Bioinformatics prediction analysis was performed by varsome based on the manual examination of the ACMG-AMP 2015 guideline.
| Gene name | Chromosome | Position | Variant name | DANN | GERP | MAF in GenomAD | CADD | Mutation Taster | SIFT | Varsome (ACMG score) |
|---|---|---|---|---|---|---|---|---|---|---|
| NOTCH1 | 9 | 139401233 | c.3836G>A, p.R1279H | 0.99 | 4 | 0.0152 | # | D | Tolerated | B1 |
| MYH9 | 22 | 36697694 | c.2517G>A, p.Q839= | 0.87 | 2.4 | 0.000223 | 16 | - | - | B |
| POLE | 12 | 133202740 | c.6494G>A, p.R2165H | 0.99 | 3.9 | 0.00468 | # | D | Damaging | B |
| IFIH1 | 2 | 163144694 | c.1046A>G, p.K349R | 0.99 | 5.9 | 0.000892 | # | D | Tolerated | B |
| FLNA | X | 153586860 | c.4551G>C, p.Gly1517= | 0.69 | 2 | 0.0000924 | 11 | - | - | B |
| MITF | 3 | 69986984 | c.366C>T, p.His122= | 0.62 | 4.6 | 0.00178 | 11 | - | - | B |
| SF3B4 | 1 | 149899116 | c.105G>A, p.Gln35= | 0.93 | 4.9 | 0.0000257 | # | - | - | B |
| ARAF | X | 47428231 | c.1046A>G, p.K349R | 0.99 | 5.9 | 0.000892 | 16 | D | Tolerated | LB2 |
| CHD6 | 20 | 40074369 | c.3813G>A, p.Glu1271= | 0.55 | 3.9 | 0.000796 | 10 | - | - | LB |
| FANCI | 15 | 89804072 | c.286G>A, p.Glu96Lys | 0.99 | 5.4 | 0.00111 | # | D | Damaging | LB |
| TSHR | 14 | 81610235 | c.1833G>A, p.P611= | 0.85 | 2.5 | GnomAD exomes = 0.0000199 | 15 | - | - | LB |
| LAMA1 | 18 | 7007194 | c.4204C>T, p.P1402S | 0.99 | 5.8 | 0.0000956 | # | D | Damaging | LB |
| PRICKLE2 | 3 | 64145697 | c.315C>T, p.S105= | 0.76 | 3.9 | 0.0000257 | 12 | - | - | LB |
| RAI1 | 17 | 17700989 | c.4727G>A, pR1576H | 0.99 | 4.7 | 0.000012 | # | D | Damaging | VUS3 |
| GPR98 | 5 | 89977185 | c.5578G>A, p.A1860T | 0.99 | 5.8 | 0.000351 | # | D | Damaging | VUS |
| SACS | 13 | 23912136 | c.5879G>T, p.C1960F | 0.99 | 5.7 | Variant not found | # | D | Damaging | VUS |
| MACF1 | 1 | 39763362 | c.2441C>G, p.S814C | 0.98 | 6 | GnomAD exomes = 0.000131 | # | D/P | Damaging | VUS |
| MDN1 | 6 | 90359793 | c.16156T>C, p.C5386G | 0.94 | 5.8 | Variant not found in GnomAD | # | D | Damaging | VUS |
| NOTCH2 | 9 | 120458867 | c.6478T>A, p.Y2160N | 0.97 | 5.7 | Variant not found in GnomAD | # | D | Damaging | VUS |
| RERE | 1 | 8421866 | c.1973C>T, p.T658M | 0.99 | 5.4 | 0.0000386 | # | D | Damaging | VUS |
| 8415606 | c.4540G>A, p.A960T | 0.99 | 5.9 | Variant not found | # | D | Damaging | VUS | ||
| Note: 1B: Benign, 2LB: Likely Benign, and 3VUS: Variant of Uncertain Significance | ||||||||||
As yet, more than 200 rare variants located in CDS of RET gene associated with HSCR have been assigned to HGMD database. The rare variants can be allocated into two groups. The null variants such as nonsense or Indels and splicing variants (mutation in canonical ± 1 or two splice sites) result in a truncated protein. The Variant of Uncertain Significance (VUS) such as most missense mutations and small in-frame deletions has appeared with less functional or nonfunctional protein features. However, it is reported that variants detected in RET gene can mainly cause the loss of function of protein in HSCR cases [24,25].
RET gene in humans contains 21 exons that encode a Receptor Tyrosine Kinase (RTKs), which has a cadherin-like extracellular domain, a cysteine-rich region, and an intercellular tyrosine kinase domain. Based on a functional assessment and location of variants, the RET mutations associated with HSCR are classified as follows: First, mutations in coding sequences that code for the extracellular domain of RET's receptor tyrosine kinase affect its transport to the plasma membrane during translation of the protein. Second, RET’s cysteine-rich domain mutations make a covalent dimerization of RTK upon ligand activation and diminish its localization at the plasma membrane. Third, mutations affecting the kinase domain destroy the activity of RET tyrosine kinase. Fourth, mutations at the C-terminal end of RET gene modified the normal signaling pathway on account of alteration in the protein-binding site. Fifth, mutations disturb RET transcription process in regulatory elements such as promoter and intron 1. These mutations potentially affect protein function and accordingly lead to HSCR [26].
In HSCR, two types of variants (rare and common) in RET have been observed, which have a significant role in the expressivity of the disease. The RET common variants have been mainly identified in the most typical forms of HSCR such as S-HSCR and sporadic forms, and the rare variants located in CDS of RET are often found in more severe conditions of HSCR like L/TCA-HSCR and familial forms. Unlike common variants, rare destructive variants usually have higher penetrance and profoundly affect the risk of developing the disease. Rare damaging variants are negatively selected, and they have to be newly (de novo) generated and potentially accumulated their population-level destructive effect [27]. Since an enormous amount of genomic data has been usually generated by NGS technology (especially WES service), planning an experiment with two or more subjects may help to determine significant disease-causing variants, and this would probably ease the difficulty of interpreting such rare variants [28].
A comprehensive understanding of the complex genetic background of the disease is a pivotal challenge requiring further efforts. In this study, a novel nonsense mutation (c.942C>A, p.Y314X) as a null variant at exon 5 of the RET gene associated with a wide range of phenotypes and incomplete penetrance of HSCR was detected. As mentioned above, this mutation, p.Y314X, may lead to a truncated protein that is excluded from a protein kinase binding domain. Nonsense variants of RET with loss of function mechanism are associated with severe pathogenic impact on HSCR phenotype. This damaging variant in the RET gene with an autosomal dominant inheritance leads to protein without function. The patient IV-V with the identified causative variant had other symptoms such as a single kidney, absence of peritoneum, and pigmentation of the face coincident with HSCR, which these manifestations could explain variable expression in this disease. However, this variant has been segregated in her mother (III-IV), grandmother (II-II), and aunt (III-V) of the patient (IV-V); these members of the family do not have shown HSCR. The single kidney and diarrhea were manifestations of subjects III-IV and II-II, respectively. Also, chronic constipation and premature ovarian failure were observed in subject III-V.
Furthermore, in the other family members (II-IV and III-XII) with chronic constipation and without HSCR, the novel variant p.Y314X was detected. This occurrence is defined by incomplete/reduced penetrance. In a meta-analysis study of RET gene in Hirschsprung disease by Puri P and Tomuschat C, it is suggested that carrying one pathogenic RET mutation maybe share a minor role in genotypephenotype correlation on its own. These variabilities are depended on genotypes at different loci and environmental factors [29,30]. As declared, HSCR is described as a complex disease influenced by numerous causative variants that might have a small solitary effect [31]. Also, two types of RET mutations, including rare coding variants (high penetrant), and common variants at the promoter and intron sequences (low-penetrant), seem to act in a synergistic mode predisposing to HSCR phenotype [32-34].
It is important to note that most HSCR cases in pedigree members without RET rare or common variants but with different phenotypes might explain unidentified mutational events in the known HSCRrelated genes or unknown genes which could act alone or in combination. In addition to modified genes, environmental and epigenetic factors can also elucidate the variable expression and incomplete penetrance in multifactorial diseases such as HSCR [35]. Ultimately, other family members (II-III, II-V, II-VII, III-VIII, and III-X) died at birth with acquired/congenital malformations such as closed anus.
Also, we identified some rare, benign, and VUS variants (heterozygous mode) in other HSCR-related genes (Table 2). As mentioned above, several variants (common and rare) with additive effects contribute to Hirschsprung disease as a complex phenotype that could be with other comorbid phenotypes. Noteworthy, additional studies on proper patients and controls along with functional studies in zebrafish models are needed to show variants’ pathogenicity.
Approximately 18% of cases with HSCR co-occur with other congenital malformations [36]. The anomalies often include genitourinary (cryptorchidism and hypospadias), cardiac (such as atrial septal defect and tetralogy of Fallot), gastrointestinal (such as intestinal malrotation), and central nervous system (microcephaly and intellectual disability) deficiencies [37].
In this study, in addition to HSCR, the family’s history showed other manifestations/malformations such as single kidney, closed anus, premature ovarian failure, and chronic constipation. However, RET mutations (rare or/and common) play a significant role in isolated and syndromic HSCR. Various authors have described that HSCR occurrence does not seem to be dependent on the RET genotype alone. Still, one or more other genes can be contributed to HSCR expression [38,39]. Further studies revealed that the standard gene networks such as the RET/GDNF signaling pathway help develop the enteric nervous system and Kidney development. Subsequently, RET mutations are recognized in various congenital abnormalities with isolated Congenital Anomalies of the Kidney or Urinary Tract (CAKUT) or HSCR [40]. For example, in a report of a patient with right renal agenesis and oligomeganephronia and total colonic aganglionosis, mutation analysis revealed a heterozygous p.S811F mutation in exon 14 of RET. In our research, the proband IV-V and her mother (III-IV) indicated a single kidney and HCSR together with identified p.Y314X mutation in exon 5.
Furthermore, based on publication data, the disease is genderdependent and the estimated penetrance is 72% in males and 51% in females [41]. Our observation in this extended pedigree shows the same thing. It seems that affected mothers (II-VI) with reduced penetrance could transfer susceptibility genes (for instance the identified nonsense variant in RET) to their alive sons. But affected males (II-IV and II-II) could not inherit susceptibility gens to their sons and just female affected fetus are born with variable expression of the disease.
This study showed that even in the case of the extended families with variable expressivity of phenotypes and the existence of incomplete penetrance, it is appropriate to consider high disease genetic complexity when counseling and performing genetic tests for disorders with a presumed multifactorial etiology. In cases similar to this family with multiple affected members and known deleterious mutation, genetic counseling should be considered part of the disease management. For families with the same scenario of having multiple affected members and segregated germline mutation, it is recommended to do prenatal diagnosis or doing the pre implantation genetic diagnosis. Investigation of other genetic variants in non-coding regions of the genome using whole-genome sequencing, assessments of their functional significance, and research on environmental or other factors would be beneficial for not only identifying penetrance and variability of the Hirschsprung disease presentations but also for providing accurate genetic counseling.
The authors extend their appreciation to the patients and their preparedness to participate in this study.
The authors declare that they have no competing interests.
Citation: Rahnavard S, Eghbali M, Saei H, Ghaffarnia R, Ardeshirdavani A, Ashjaei B, et al. (2022) Assessment and Documentation of Indigenous and a Novel Nonsense RET Variant in an Extended Hirschsprung Iranian Pedigree with Reduced Penetrance and Variable Expression. J Hepatol Gastroint Dis. 8: 216.
Received: 24-Aug-2022, Manuscript No. JHGD-22-19025; Editor assigned: 29-Aug-2022, Pre QC No. JHGD-22-19025 (PQ); Reviewed: 09-Sep-2022, QC No. JHGD-22-19025; Revised: 13-Sep-2022, Manuscript No. JHGD-22-19025 (R); Published: 19-Sep-2022 , DOI: 10.35248/2475-3181.22.8.216
Copyright: © 2022 Rahnavard S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.