Fungal Genomics & Biology
Open Access
ISSN: 2165-8056
ISSN: 2165-8056
Review Article - (2013) Volume 3, Issue 2
Keywords: Fungal diversity, Next-generation sequencing, ITS,Environmental genomics, Metagenomics
AD: Atopic Dermatitis; AFTOL: Assembling the Fungal Tree of Life; BLAST: Basic Local Alignment Search Tool; BOLD SYSTEMS: Barcode of Life Data Systems; CAMERA: Community Cyberinfrastructure for Advanced Microbial Ecology Research & Analysis; CloVR: Cloud Virtual Resource; DDBJ: DNA Data Bank of Japan; DFVF: Database of Fungal Virulence Factors; DOE/US: United States Department of Energy; EMBL: European Molecular Biology Laboratory; Fungi DB: Fungal and Oomycete Genomics Resources; GI: Gastrointestinal; GOLD: Genomes Online Database IMG/HMP M: Integrated Microbial Genomes with Human Microbiome Project; IMG/M: Integrated Microbial Genomes with Microbiome Samples; INSD: International Nucleotide Sequence Databases; ITS: Internal Transcribed Spacer; ITS1: Ribosomal Internal Transcribed Spacer 1; ITS2: Ribosomal Internal Transcribed Spacer 2; ITSone DB: Collection of the fungal ribosomal RNA Internal Transcribed Spacer 1; LSU: Large Subunit; MEGAN: MEtaGenome Analyzer; MG-RAST: Metagenomics RAST Server (RAST: Rapid Annotation using Subsystem Technology); NCBI: National Center for Biotechnology Information; NGS: Next Generation Sequencing; OUT: Operational Taxonomic Unit; PCR: Polymerase Chain Reaction; PHYMYCO-DB: Fungi phylogenetic sequences database; PlutoF: Web Based Workbench for Ecological and Taxonomic Research; QIIME: Quantitative Insights into Microbial Ecology; RDP: Ribosomal Database Project; SNPs: S i n g l e Nucleotide Polymorphisms; SSU: Small Subunit; UNITE: Fungal rDNA; ITS: sequence database; WGS: Whole Genome Shotgun
Fungal diversity studies have traditionally relied on morphologic and other phenotypic characteristics, and these were for many years the main criteria for fungal classification [1]. However, due to the instability of morphological traits, the existence of intermediate forms and the phenotypic overlap between different taxa [2], these methods alone do not enable a reliable identification of fungi at lower taxonomic levels [3], even at the light of modern techniques [4]. Molecular taxonomy has partially solved this problem, allowing for better species classification of fungi [2,3,5-7], even though some authors believe the ribosomal DNA sequence alone is not inherently superior to morphological taxonomy [8]. Thus, the use of hybrid approaches has been the methodological choice in many studies [6,7].
The development of whole shotgun genome sequencing in the late 1990s boosted eukaryote genomics, and a main landmark in this field was the sequencing and assembly of the first fungal genome: that of the yeast Saccharomyces cerevisiae[9]. The number of complete fungal genomes sequenced has increased considerably since then (more than 2,400 fungal genome projects were registered at GOLD (Genomes Online Database) at the time of writing this manuscript), together with the number of fungal sequences in environmental DNA databases, as reviewed herein. This increase reflects the advances in the area of metagenomics (also referred to as environmental genomics or community genomics) - a culture-independent approach for the study of genomes collectively recovered from the environment [10-13]. The term “environment” here is used in a broad sense, comprising all typical environmental compartments (air, soil, sediments, continental, oceanic, and ground water) as well as the external and internal surfaces and microenvironments within macroorganisms [14].
The idea of collectively analyzing microbial communities and directly assessing their genomes is not new [15-17]. It can be traced back to a period long before the first organism had its genome completely sequenced [18] or the term “metagenomics” was officially coined and first appeared in a scientific work [19].
Following the pioneer work by Jo Handelsman and co-workers, in which soil metagenomic clone libraries were built and screened for biological activity [19], other similar studies were conducted using the shotgun approach [20,21]. The first reconstruction of multiple genomes directly from a natural sample was achieved [22], and the study conducted by Venter and collaborators in the Sargasso Sea was another important landmark [23]. In the later study, the authors identified over 1.2 million new genes and 148 novel bacterial phylotypes [23]. This represents a shift in magnitude of metagenomic studies and is a first clear demonstration that whole shotgun sequencing approach can be applied to large-scale phylogenetic surveys of uncultured organisms in environmental samples [24].
In 2004, 454 Life Sciences lauched the first commercial next generation sequencing (NGS) platform based on pyrosequencing [25]. Soon after that, Solexa (purchased by Illumina in 2007) launched the Genome Analyzer and Applied Biosystems launched the solid platform. In spite of the different chemistry used, these NGS platforms all enabled a massive parallelization of sequencing, which considerably scaled up the throughput [26]. In addition, they eliminated the need for cloning environmental DNA, thus reducing the bias often associated with this step [27,28]. NGS technologies proved to be robust enough for shotgun sequencing and assembly of whole genomes, in spite of the substantially shorter reads they generate [26].
The first metagenomic study using NGS was that by Edwards and co-workers on deep mine microbial ecology [29]. They used pyrosequencing to study two different samples in the Soudan Mine (Minnesota, USA) and they observed differences in the metabolic potential of the microbial communities in these environments. This study employed a systemic approach, integrating biology, chemistry and geology, which were an important advancement in modern microbial ecology, bringing our view of the microbial world to a more holistic perspective.
Because of the high speed, low cost, and significant technical advantages [29], NGS has been extensively used in metagenomics in the past decade. As a consequence, the number of metagenome projects has increased exponentially [30]. An unprecedented number of datasets became publicly available, but most of the data are related to prokaryotic microbial communities [31-33]. Therefore, the eukaryotic component of the communities and fungal taxa in particular, still remain considerably underrepresented [34]. Attempts to focus metagenomic studies in eukaryotes included the use of special sampling procedures [35-37] and whole genome amplification [34,38].
In this review, we analyze the current knowledge on fungal diversity, the main methodological advances that took place over the past decade, and the main challenges and future directions in this area.
Estimates on fungal global species richness have ranged from conservative (611,000) [39] to more optimistic ones (9,900,000) [40]. However, fungal diversity is likely underestimated because these calculations do not consider fungi in non-soil habitats or fungi not associated with plants, as well as the fact that fungal diversity in the tropics is arguably much larger than previously thought [41,42] for a review.
In the following sections we describe a collection of published research results on the study of fungal diversity in the natural environment (i.e. water, air and soil).
Water
Approximately 71% and 0.8% of the Earth’s area is covered by saltwater and freshwater, respectively [43,44]. These ecosystems are regarded as the largest bioproductive resources, and oceanic microorganisms are responsible for up to 98% of primary production [45].
Fewer studies have been conducted in freshwater than in saline ecosystems and these are frequently focused on prokaryotic diversity [44]. However, fungi are ubiquitous and play major roles, primarily as parasites and saprotrophs, in most aquatic environments [46,47]. Early microbial diversity studies in aquatic environments were based on clone library analysis, a labor-intensive and expensive approach [19-23,48]. However, NGS-based approaches have been preferred in recent years [49].
The fungal abundance in aquatic ecosystems can vary greatly, and the environmental community profile can be influenced by sampling methods [50] and environmental conditions, including anthropogenic activity [43]. A study conducted in Sichang Island (Thailand) using pyrotag sequencing compared the metagenomes of two coastal areas with similar oceanographic positions, which differ in bay geography and the extent of municipal disturbances [43]. Among the 18S rDNA sequences, it was found that fungi, mainly the Basidiomycota, accounted for around 75% of the organisms detected in Tha Wang Bay. Constrastingly, fungi were ten times less abundant in Tham Phang Bay [43], in which over 80% of the 18S rDNA sequences were assigned to Metazoa (specifically to Brachiopoda and Mollusca). Another comparative metagenomics study, conducted in the Sea of Marmara, found out that Fungi and Metazoa represented 30% of the total sequences obtained from sediment samples, but these organisms were poorly represented in the bathypelagic planktonic samples [51].
An important initiative towards understanding the dynamics and ecology of fungi in freshwater ecosystems is that by Monchy et al. [52]. The authors studied two French lakes, Pavin and Aydat, employing two approaches: a classical, consisting of cloning/sequencing of the ITS region, and the pyrosequencing of 18S rRNA hypervariable regions. The first approach allowed for the attainment of 146 (Lake Pavin) and 143 (Lake Aydat) sequences, corresponding to 46 and 63 OTUs, respectively. In Lake Pavin, half of the OTUs identified match to Fungi, mainly spread in the Chrytridiomycota (17), Ascomycota (7) and Basidiomycota (1) phyla. In Lake Aydat, one-third of the OTUs corresponded to Fungi, including 10 Chytridiomycota, 8 Ascomycota and 1 Basidiomycota. Within all the identified OTUs, only two Fungi were common to both lakes: one Ascomycota and one Chytridiomycota. The pyrosequencing approach yielded 42,064 (Pavin) and 61,371 (Aydat) reads, of which 12- 15% and 9-19% reads were assigned to fungi in Lakes Pavin and Aydat, respectively. Chytridiomycota members were also dominant among these reads. While the later technique provided a general overview of the eukaryote diversity, unveiled rare species, and gave quantitative information about the OTUs, the classical approach attributed to each sequence a precise taxonomical position and identified potential new clades. Finally, one of the major findings of the study was that the two lakes exhibited dissimilar spatial distributions, homogenous for Lake Pavin and heterogeneous for Lake Aydat, which may be related to their particular characteristics.
Clone libraries (n=100) constructed for RNA (cDNA) and DNA sequencing using the 3730 Genetic Analyzer (Applied Biosystems) were used by Rao et al. [53] to recover the fungal biodiversity in freshwater sediments from a subtropical forest. The two above-mentioned methodologies plus a cultivation-based approach were selected in order to compensate the bias each methodology carries by itself. The results obtained for the three approaches revealed Anguillospora furtiva as the dominant fungus. This taxon comprised 85–86% DNA libraries, 90– 91% RNA libraries and was cultivable from all samples. The remaining taxa were phylogenetically diverse and stretched over Ascomycota, Basidiomycota and subphyla incertae sedis. The data obtained from this study indicate the less abundant taxon in an environment may be subjected to greater bias when a single approach is used to estimate diversity.
Air
The late 20th century saw an increase in studies investigating the effects atmospheric particulate matter such as dust, industrial pollutants and microorganisms could be inducing on human health, agriculture, and climate [54].
A revisiting of aerobiology and technological advancements has paved the way for various studies in an attempt to characterize the effects of fungal aerosols [55,56]. As the predominant microbial group found in air is bacteria, this was the focus of the majority of early studies. Many of these studies were conducted in hospitals, as immunocompromised patients are the most vulnerable to infection demographic of society. Previously undescribed pathogenic fungi were identified in these studies, as well as circumstances where a usually harmless fungal invasion could result in a life-threatening situation [57,58]. More work followed regarding effective preventative and monitoring strategies [59,60], along with the improvement of sample collection methods [61-64]. However, relatively little was known about the composition and abundance of airborne organisms until much more recently.
Metagenomics came to the fore of aerosolic research when the first airborne metagenome was produced in a study by Tringe et al. [65]. The researchers attempted to determine the composition of airborne microorganisms in two indoor shopping centers in Singapore. Air samples were collected through a vacuum mechanism and aerosolic organisms caught by two different types of filters. DNA was extracted from the entire microorganism pool; the 16S rDNA genes were amplified by PCR; and then sequenced using an ABI3730 sequencer. In addition, small insert libraries were built and shotgun sequenced. That study identified that indoor populations were distinct from those commonly found outdoors and confirmed that the majority of biogenic aerosols were of prokaryotic origin, with eukaryotes comprising a smaller but significant proportion (between 0.26 and 2.0 % of the shotgun reads).
Fungal metagenomic studies from fine and coarse air filter samples were conducted by Fröhlich-Nowoisky et al. [66] using a similar approach, based on the amplification and cloning of the ITS region, followed by Sanger sequencing. The ITS1 and ITS2 regions were used for analysis and taxonomic attribution. That study found that the diversity of fungal species in air was similar to that present in soil or on plants. Known human allergens and pathogens were found to be slightly more prevalent in the fine particle samples, whereas plant pathogens were mostly found in the coarse samples. Fine particles have a much longer residence time in the atmosphere, thus studies of biogenic aerosol effects on human health and climate are primarily focused on fine particle aerosols. This study revealed that diversity of fungal species in the atmosphere is much higher than previously reported [67-69].
Qian et al. [70] also used a NGS-based metagenomic approach for analyzing airborne bacteria and fungi in occupied classrooms. Although this study was primarily aimed at calculating total amount and emission rates of aerosolic fungal spores, it revealed that concentrations increased in an occupied space as opposed to unoccupied. This was thought to be due to emissions from humans. It was also noted that the largest increase in concentration occurred in the largest particles (>9 μm) whereas the smaller particle size concentration increased only subtly. Significantly large increases were seen in multicellular spores such as Alternaria and Epicoccum species.
Another interesting study by Amend et al. [71] used indoor samples taken from 72 locations globally in an attempt to ascertain whether global or indoor factors determine indoor fungal compositions. Buildings on six continents were the collection sites and samples were pooled from a range of areas in these buildings deemed to be “accessible”, “infrequently accessed” and “inaccessible”. DNA was extracted from the sample pool, the ITS1/ITS2 and LSU regions amplified by PCR, and then multiplexed in a sequencing reaction on the 454 GS FLX platform. Samples were classified into 4,473 OTUs (operational taxonomic units), through bioinformatic analysis, only 31 of which were represented in more than half of the samples. The results revealed there is a strong correlation between fungal diversity and latitude. Also, it suggested populations in indoor environments in temperate zones are more diverse than those in tropical sample sites. The authors also concluded that there is no significant relationship between factors such as building materials or content and fungal composition.
As evidenced by the small number of airborne metagenomes produced, this is a relatively unexplored topic, even more so from the perspective of fungal aerosols. However, given that around 100,000 species of fungi have been described to date by taxonomists, and a significant number of which transmit spores through the atmosphere [72], it is clear that this is an area that requires further investigation. Given that we are already aware of many human diseases, such as asthma and aspergillosis, which are caused or exacerbated by aerosolic fungal spores [73,74]; the relevance of these studies cannot be refuted. Research in this area is developing quickly and it can benefit from the newest tools and well suited metagenomic approaches available today.
Soil
Soils are among the most diverse and densely populated microbial habitats on Earth, harboring high taxonomic and functional fungal diversity [75,76]. Studies focused on soil mycobiome have revealed fungal diversity to be influenced by soil stratification and vegetation coverage [77,78]. Analysis of litter and organic horizons from spruce (Picea abies) forest in Central Europe [77] and plantations from the Morvan Mountains in France [78] identified Basidiomycota and Ascomycota as the prevalent fungal sequences. Basidiomycota accounted for 65% and 28% of OTUs in soils from oak plantations and spruce plots, respectively [78]. Sequences assigned to the Glomeromycota were identified in a low proportion (2.24%) in the first environment and were not found in the second [78]. This spatial heterogeneity seems to be determined by the host tree and soil organic matter composition. The ITS region amplification in this study was designed for Dikarya, and this fact may explain the low occurrence of Glomeromycota in these datasets. In another study, Ascomycota was the most prevalent fungal kingdom, accounting for 36.7 to 93% of all OTUs, for most samples from different ecosystems across Italy and France [79].
Uroz et al. [80], whilst evaluating the microbial communities in soil of a spruce plantation (France) using a combined 454 and Illumina sequencing approach, reported that only 0.2% of the annotated reads have a significant match to fungi, and these are more abundant in the organic horizon than in the mineral horizon of the soil.
Arfi et al. [81] used internal transcribed spacer (ITS) rDNA pyrotag sequencing to evaluate the fungal diversity in anoxic-sulfidic sediments in mangrove soil and found that 50% of the reads belong to Basidiomycota, mainly to the Agaricomycete class. Sistotremastrum, a saprobe fungus usually found in association to rotten wood [82], is the dominant fungal genera in this environment. Moreover, many ubiquitous plant pathogens and degraders, such as Alternaria, Galactomyces, and Penicillium, were detected.
Through the use of a metagenomic approach, it has been shown that agricultural practices affect fungal diversity in soil [83]. Rascovan et al. [84] used 454-FLX Titanium chemistry to perform a deep sequencing of the Argentinean Pampean soil metagenome (36 shotgun libraries totaling 17.8 million reads or 7.7 GB). In this study, only 1% of the reads were identified as Eukarya, and among these, 27% are of fungal origin. This high quality metagenomic dataset (PAMPA datasets) has per-sample associated metadata and is publicly available [84].
The microbial community associated with animal or plant hosts, or with specific tissues or organs in these organisms, is generally known as “microbiota”, and the collective genome of the microbiota is referred to as “microbiome” [85]. Accordingly, the fungal component of these communities constitutes the so-called “mycobiota”, and their genomes are referred to as “mycobiome” [79,86]. Below, we discuss some studies on the fungal diversity in the host- associated environment.
Plants
The diversity of endophytes was recently reviewed by Porras-Alfaro and Bayman [87]. These authors point out that one main challenge for the metagenomic study of endophytic fungi is the technical limitation of separating microbial from host DNA. Plant DNA is much more abundant than fungal DNA, making it difficult to isolate and sequence the fungal metagenome at high coverage.
Unterseher et al. [88] addressed the suitability of species abundance models in three groups of plant-associated fungal communities – phyllosphere, ectomycorrhizal, and arbuscular mycorrhizal fungi, using 454 sequencing data. The authors pointed out that there are no one-size-fits-all solutions, and highlighted several challenges. For instance: spatial and temporal alternation of life cycles may strongly affect composition of fungal communities in soil and phyllosphere; and in order to unravel the true richness of microbial communities, it may be more important to invest in intensive sampling rather than intensive sequencing.
Danielsen et al. [89] studied the diversity of fungi in soil and roots of three different poplar. By 454 pyrosequencing targeting the rDNA internal transcribed spacer 1 (ITS1) region, the authors demonstrated that fungal species and family richness in the soil is surprisingly high in this simple plantation ecosystem. Their data suggest saprophytic, pathogenic, and endophytic fungi are the dominating groups in soil, whereas ectomycorrhizal fungi are dominant in roots (87%). Also, according to their results, arbuscular mycorrhizal diversity is higher in soil than in roots.
454 pyrosequencing was also used to assess the fungal diversity and spatial distribution of phyllosphere in European beech (Fagaceae) [90]. The authors observed highly diverse fungal assemblages, with high proportions of generalist and cosmopolitan fungi, and suggested that the genetic variation of trees is a possible determinant of phyllosphere fungal communities.
As highlighted by Porras-Alfaro and Bayman [87], plant-associated fungi, endophytes in particular, are an underexplored source of biomolecules, and we believe that metagenomic studies can add an important contribution to the efforts towards their culturability.
Invertebrates
Fungus-invertebrate associations are among the most interesting chapters of ecology. Some fungal taxa contribute to invertebrate nutrition [91], while others have a parasitic mode of life [92]. More than 750 species of fungal invertebrate pathogens were described [93], and some are regarded as potential useful biocontrol agents [94,95].
Several studies point to the existence of multiple and diverse origins for insect parasitism within the fungal kingdom, and shifts to trophic specialization on insects seem to have evolved in all phyla at least once [92]. Among the Basidiomycetes, a group that have evolved many different symbiotic associations with animals and plants, the Septobasidiales are the only large group that are obligatory parasitic of insects [94]. Other “hotspot”-taxa for insect parasitism are the Entomophthorales and Eccrinales (Zygomycota), the Laboulbeniales and Hypocreales (Ascomycota) [96,97].
Nematophagous fungi are a complex group of organisms, and they can be classified according to their mode of infection [98]. Cheng et al. [99] used a metagenomic approach to investigate the role of nematode microbiome in xenobiotics detoxification. Baquiran et al. [100] used a ribosomal rDNA targeted approach to study the microbiome associated with the nematode Acrobeloides maximus. However, none of these studies focused on nematode-associated fungal communities.
The mycoflora associated with living and dead molluscs were found to comprise zoosporic fungi [101], and the species richness is believed to be correlated with the composition of the molluscs mucous cover and stressogenic factors, such as water temperature and nutrient content, which may affect the invertebrate resistance to infection.
Fungi were shown to be the most abundant group of organisms associated to the Caribbean coral Porites astreoides (a coral holobiont), outnumbering bacteria more than 5 times [38]. The Ascomycota (mainly Sordariomycetes) accounted for the majority (93%) of the fungal sequences, but Basidiomycota and Chytridiomycota were also detected. The taxonomic composition of the coral-associated Ascomycetes, as assessed through 18S rDNA amplicon sequencing, is consistent with the classification of functional genes.
For over a century, denitrification and ammonification have been considered processes performed by prokaryotes, but studies have shown that fungi are able to participate in these cycles [102]. Fungal genes obtained from coral holobiont by metagenomic approaches [38] related to the carbon and nitrogen metabolism cycles, are suggestive of the participation of these organisms in those biogeochemical pathways.
Newton et al. [103] discussed the usefulness of invertebrates for hypothesis-driven microbiome research. The use of genetically amenable model organisms can help shed light to the complex relationships involved in the hologenome concept [104].
Non-human mammals
The mammalian gastrointestinal tract (GI) is one of the most complex microbial ecosystems. Gut microbiota influences host health by stimulating the immune system, providing competitive exclusion, and nutritional benefit to the host [105]. The healthy state of animals seems to be fine-tuned with their microbiome composition. Indeed, GI disease in dogs and cats are correlated to a microbial imbalance [106].
Tun et al. [107] and Swanson et al. [108] studied the gastrointestinal microbiota of feline and dog fecal samples, and found only a low proportion (around 0.02%) of fungal sequences. Suchodolski et al. [109] found, in a study that evaluated fungal presence by sequencing the ITS DNA of healthy and diseased dogs, a higher amount of sequences (76%) in the animals with chronic enteropathies than in healthy ones (61%). As much as 51 different fungal phylotypes were identified among the samples and were classified as Ascomycota (32 phylotypes) or Basidiomycota (19 phylotypes). These results indicate high prevalence and diversity of fungal DNA in the small intestine of both samples. Other studies evaluating healthy and diseased canine and feline fecal samples also identified the majority of sequences belonging to the phyla Ascomycota (>90%). Saccharomycetes have also been reported to occur in fecal samples of these animals [110,111].
Rumen is an interesting ecosystem with a high microbial population density, diversity, and complexity of interactions. Due to the presence of unique microorganisms, rumen is effective for the conversion of plant cell wall biomass to microbial proteins, short chain fatty acids, and gases. Rumen microbiota is dominated by bacteria, but a variety of anaerobic protozoans, archaea, and fungi [112] can also be found [113]. A study of the rumen microbiome of Surti Buffalo by Singh et al. [114] found that eukaryotes represent 10 to 17% of sequences in all samples, with most belonging to fungal and metazoan groups. Sequencing of the 18S rRNA gene of fungi from bovine rumen suggests that the compositional characterization of this microbiome is incomplete with several novel fungal taxa being discovered (from the 71 total fungal OTUs identified, only 53 grouped near a previously deposited sequence) [115].
The survey of microbiota communities associated to mammal’s GI tract and rumen and analysis of their correlations suggested that much of microbial diversity (mainly fungal) is yet to be discovered. Studies using metagenomic approaches certainly contribute by revealing interactions and pathways useful for better understanding as to how microbial community structure and function can affect host health and disease. Also, it is expected that the detailed comprehension of the conversion of plant cell biomass into simpler compounds by microorganisms present in the rumen will encourage the development of new biotechnological processes, such as enzymes for the biofuels industry [116].
Humans
The number of cells in the microbiome of one human individual outnumbers the human cells by ten times, while the metagenome of the human microbiome has at least 100 times as many genes as the human genome [117]. Fungi are believed to play an important role in human microbial community stability, thus affecting human health and disease [118,119]. The mycobiota of the skin, gut and other mucosal sites has gained more attention in the past few years, as discussed below.
Skin: Skin microbiota, especially commensal microorganisms, play an important role in modulating immune response and maintaining epithelial health [120]. Factors known to affect the distribution and diversity of microorganisms on skin include: sebaceous gland density, moisture content, temperature, exogenous environmental factors, and host genetics. Consistent with other environments, samples of human cutaneous fungal microbiota from both diseased and healthy individuals showed Ascomycota and Basidiomycota to be the predominant phyla in varying quantities.
In an rDNA clone library-based study aimed at comparing the skin fungal microbiota of patients with atopic dermatitis (AD) and healthy subjects, Zhang et al. [121] demonstrated that genus Malassezia is predominant in both cases. However, the non-Malassezia yeast microbiota form a more diverse group on the patients with AD than on healthy individuals.
Park et al. [122] investigated the fungal communities associated with dandruff on the human scalp using high throughput sequencing of ribosomal 26S amplicons (over 65,000 454 sequence reads with average size of 440 bp). This study identified differences in the abundance of phyla and species between healthy and affected individuals. According to their results, Acremonium is a common Ascomycete fungus on both healthy and dandruff-affected scalps. Among the Basidiomycota, Cryptococcus is the predominant genus on healthy scalps, while Filobasidium spp. is the most abundant on dandruff-affected ones.
Findley et al. [118] also used 454 pyrosequencing to study 14 skinsites from ten healthy volunteers, and observed that there is higher fungal diversity between body sites than between individual subjects. Remarkably, while 11 core-body and arm sites show little diversity at the genus level, representing stability, significantly higher fungal diversity was observed on three foot sites, both within and between individuals. The authors suggest that ecologically unstable areas, such as the foot, are also the ones more frequently affected by disease. The combined analysis of bacterial and fungal communities indicated that physiological state and skin topography are the main factors influencing the composition of these communities. It also shed light on how the interactions between pathogenic and commensal fungal and bacterial communities relate to skin diseases.
Gut: Microbial eukaryotes represent an important component of the human gut microbiome, playing either beneficial or harmful roles. Some species are commensal or mutualistic, whereas others are opportunistic or parasitic [123]. The eukaryotic component of the human gut microbiome probably remains relatively unexplored because eukaryotes are less abundant than bacteria or because they are not as widely studied using culture-independent methods [124,125]. The later hypothesis is supported by the results found by Hamad et al. [126], who studied the eukaryotic microbiota of a single fecal sample from a healthy African male using both culture-dependent and cultureindependent methods.
When investigating the relationships of diet with fungi and archaea of the human intestinal microbiome, Hoffmann et al. [127] suggested a syntrophic association. In this relationship, Candida would degrade starch into simpler sugars, which could then be metabolized by bacteria such as Prevotella and Ruminococcus. Fermentation byproducts would then be consumed by Methanobrevibacterwith the subsequenct production of CO2 and/or CH4.
Schwartz et al. [128], aware of the importance of understanding the mutualistic relationship between gut microbiota and the host, studied the host transcriptome and microbiome in breast-fed and formula-fed infants. They demonstrated that diet in the early neonatal period affects gut colonization and expression of genes associated with the innate immune system.
Oral: The oral mycobiome in healthy individuals was studied in 2010 by Ghannoum et al. [129], using a novel multitag pyrosequencing approach. In this study, the authors showed high fungal diversity among different individuals and four pathogenic fungal genera, Candida, Aspergillus, Fusarium, and Cryptococcus, were predominant. While the abundance of Candida in these samples is not surprising, it is higher than previously reported by studies using culture-based methods [130,131]. The presence of Aspergillus, Fusarium and Cryptococcus was unexpected, as these fungi have not been reported to be indigenous to the oral cavity. Moreover, 60 fungal genera usually found in the environment were also identified.
Other mucosal sites: Recently, Chaban et al. [132] characterized the organisms present in the upper respiratory tract of a range of individuals with H1N1 infection. 454 pyrosequencing of amplicon libraries of the cpn60 universal target revealed that fungi represent a small proportion (0.1%) of the sequence reads.
To date, little is known about the fungal microbiota of the lungs. In a recent study by Charlson et al. [133], the mycobiome of the lungs in select healthy and lung transplant recipients was analyzed. In the bronchoalveolar lavage of healthy volunteers, there was minimal fungal ITS amplification, while Candida, Aspergillus and Cryptococcus species were present in lung transplant recipients. Because all of the transplant recipients had been treated with antibiotics and immunosuppressants, this first study of the lung fungal microbiome supports the notion that host defense, and perhaps some sort of bacterial microbiome-mediated resistance mechanisms, play a major role in keeping fungal colonization low in the lungs.
As exemplified throughout this review, two main methodological strategies have been used in the study of microbial diversity. The first one is referred to as targeted metagenomics and is based on the PCR amplification and sequencing of one or more molecular markers [134]. Even though this approach does not involve direct metagenome sequencing, it is very informative with regard to the microbial community composition and it is becoming increasingly useful in the new field of quantitative metagenomics [135]. The second approach is the random shotgun sequencing of the metagenome, named shotgun metagenomics [10,11,29,134] which allows for the evaluation of the whole metagenome, and thus the assessment of the community structure and gene content [136].
The use of the ribosomal RNA gene and its variable regions as taxonomic markers for the classification of prokaryotes is well established [137-141] and the advantages and disadvantages of using them in the taxonomic profiling of metagenomes have been discussed [142,143]. A considerable effort has also been made to establish similar universal molecular markers for fungal taxa [144,145].
Fungal molecular taxonomic studies were intensified in the early 1990s [146] and have relied heavily on the analysis of the nuclear ribosomal gene cluster, which comprises the 18S or small subunit (SSU), the 5.8S subunit, and the 28S or large subunit (LSU) genes [147-151]. However, while the SSU and LSU are very efficient in the differentiation of high taxonomic levels, they are not as good for intraspecific resolution. The ITS1 and ITS2 regions were shown to be more suitable markers for fungal phylogenetic studies due to their high degree of interspecific variability, conserved primer sites, and multicopy nature in the genome [152]. The utilization of the ITS regions as universal DNA barcode markers for fungi was formalized by Schoch and collaborators [153]. This study tested the potential of four markers (ITS, LSU, SSU, and rpb1), with ITS having superior species resolution for a broad range of taxonomic groups. The ITS region was also shown to be useful for intra-specific differentiation.
Vialle et al. [154] also tested the potential of 14 mitochondrial genes encoding subunits of the respiratory chain complexes for Basidiomycota DNA barcodes. They observed that some candidate genes have the in silico potential for barcoding. However, when biological validation was conducted none had a better taxonomic resolution than the ITS marker. There are also other molecular markers that are used to study fungal phylogenetic diversity, such as EF1-α (tef1), β-tubulin (tub1, tub2), actin (act1), or RNA polymerase II subunits (rpb1, rpb2) [155-158].
The quality of metagenomic DNA is determinant for the success of both targeted and shotgun studies [159,160]. Much has been done to improve sampling. Sample size calculation and design, as well as standardized methods for the isolation of high quality DNA have been proposed and validated, including the use of commercial kits as tailormade solutions for different types of samples. In spite of this, there is still room for significant development in this area, as evidenced by the low representativeness of fungi in the main metagenomic databases compared to bacterial sequences. The output of fungal metagenomic studies is dependent on the methodological strategy used, but also on the computational tools chosen for sequence analysis. Not surprisingly, bioinformatics has quickly turned into one of the main challenges and a bottleneck in metagenomic research [161].
Figure 1 summarizes the current methods in metagenomic data management and analysis. Some of the pipelines mentioned therein were designed to accept long reads, such as those derived from Sanger and 454/Roche sequencing, as input. Others, such as QIIME [162] and MG-RAST [163], were designed to directly accept short-read data from the Illumina/Solexa, SOLiD, and Ion Torrent/Applied Biosystems platforms.
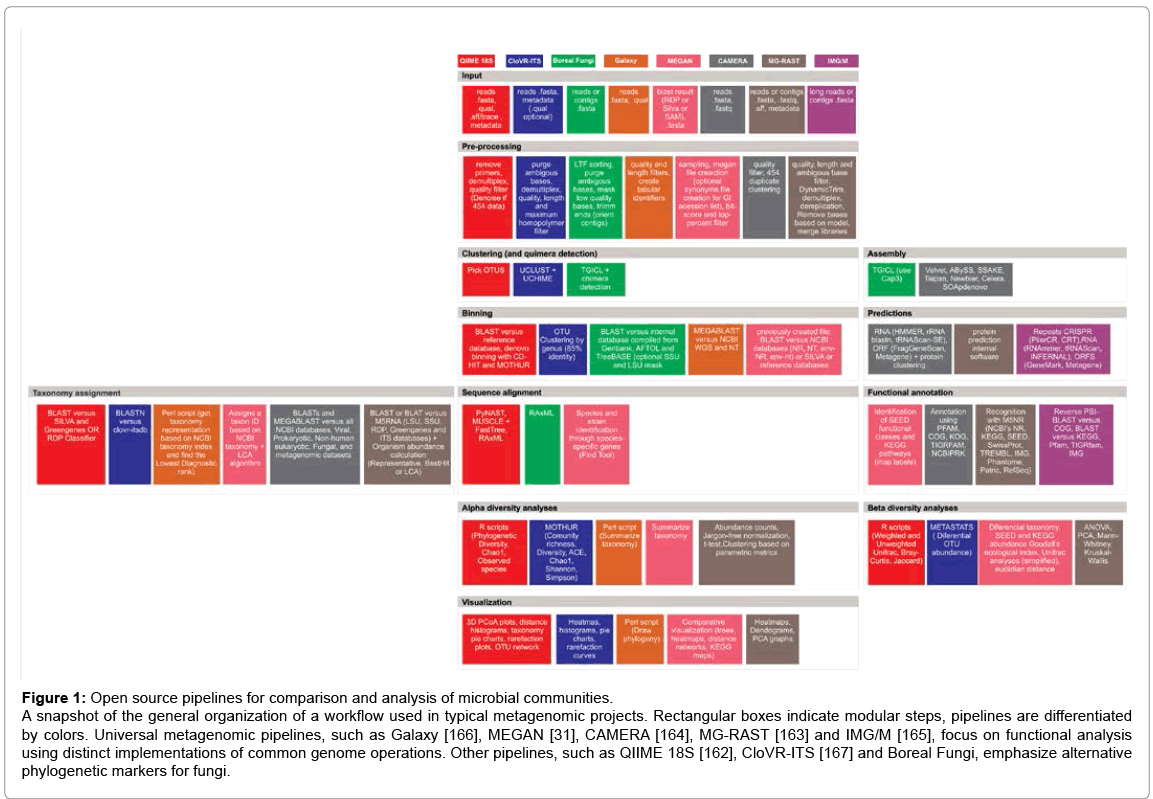
Figure 1: Open source pipelines for comparison and analysis of microbial communities.
A snapshot of the general organization of a workflow used in typical metagenomic projects. Rectangular boxes indicate modular steps, pipelines are differentiated by colors. Universal metagenomic pipelines, such as Galaxy [166], MEGAN [31], CAMERA [164], MG-RAST [163] and IMG/M [165], focus on functional analysis using distinct implementations of common genome operations. Other pipelines, such as QIIME 18S [162], CloVR-ITS [167] and Boreal Fungi, emphasize alternative phylogenetic markers for fungi.
Besides data type, several factors must be taken into account when deciding on which workflow to use. Firstly, the use of high quality information and associated metadata will improve the systemic understanding of the environment. Secondly, pipelines such as CAMERA [164], MG-RAST [163], and MEGAN [31], perform interactive analysis and comparison of the taxonomical and functional content of shotgun and amplicon datasets. MG-RAST uses FragGeneScan (FGS) and a similarity search of ribosomal RNA identification against a nonredundant integration of the SILVA, Greengenes and RDP databases. CAMERA uses MetaGeneAnnotator (MGA), while IMG/M [165] employs a combination of tools, including FGS and MGA. In IMG/M genes are predicted and putative gene functions are assigned. This annotation can be performed on the entire community and relies on unassembled reads or short contigs. The Galaxy pipeline [166] is suitable for a generic taxonomic representation, in which the reads are aligned (megablast) only against the contents of NT and WGS databases (NCBI). MEGAN is used for visualizing annotation results derived from BLAST searches in a functional or taxonomic dendrogram, and also makes analysis of particular functional or taxonomic groups visually easy. Kosakovsky and collaborators [166] directly compared MEGAN with Galaxy, and concluded the results produced were nearly identical. QIIME and CloVR [167] are applicable for the 16S, 18S, nihH, ITS genes and viral metagenomes. Although several function-oriented reference databases are available, none cover all biological functions, and their function classification system does not follow a same standard [168]. In this context, a framework that allows wide visualization and merges interpretations, such as MG-RAST and IMG/M, seems to be more informative.
Thirdly, the length of the reads has to be taken into account. MGRAST requires 75 bp or longer reads for gene prediction and similarity analysis that provides taxonomic binning and functional classification. IMG/M requires the use of assembled contigs for the analysis of more complex genetic elements. However, when studying a complex community with low sequencing depth or coverage, it is unlikely that many reads will cover the same fragment. In this case, the use of the short sequence setting at the filter homology parameters would allow better recovery from the library [169].
Furthermore, data-processing hardware requirements can present another challenge for the analysis of large datasets. In order to address this, QIIME, CloVR, and Boreal Fungi pipelines can ease computational requirements by clustering near-identical reads, resulting in faster execution. Finally, the use of a web interface to perform comparisons using a number of statistical techniques applied to stored computational results is desired. This feature is present in IMG/M and MG-RAST and is useful in beta diversity analyses, for instance, enabling comparison of novel metagenomes and re-analysis of all datasets.
Centralizing resources and standardizing annotations are relevant to address questions of microbial ecology, evolution, and diversity [170]. As studies become increasingly more complex and comprehensive, the utilization of correct tools for analysis, storage, and visualization is fundamental to ensure the best outcome from metagenomics.
Many databases are available for fungal taxonomic studies. FungiDB [171] is a resource for genomic and functional genomic data across the fungal kingdom. Its current release (FungiDB 2.3, June 2013) contains 52 complete genomes (46 from Fungi and 6 from Oomycetes), representing almost a threefold increase from its first release in 2011. Another important database used for fungal phylogenetic analyses is PHYMYCO-DB [172]. This is a manually curated bank of over 10,000 sequences from SSU rRNA and EF1-α markers extracted from Genbank (NCBI) and subjected to quality control.
For the rDNA ITS region there are a number of publicly available databases. UNITE is a fungal rDNA ITS sequence database [173]. Its main purpose is to improve the identification of fungal sequences in environmental samples [173,174]. It contains 7,802 ITS sequences from 2,120 species and all fungal ITS sequences from the International Nucleotide Sequence Databases (INSD: NCBI, EMBL, DDBJ). This represents a total 342,448 sequences at the time this manuscript was finished. UNITE also offers a tool called PlutoF, with which users can store field data, manage sequences, and conduct analysis. Other ITS databases are ITSone DB [175] and ITS2 Database [176-179]. The former is specific for fungal taxonomy while the latter includes sequences from eukaryotic taxa. Currently, the ITSone DB has 405,433 ITS1 sequences, while the ITS2 Database (version 3.0.13) has 288,044 ITS2 sequences divided into the main fungal phyla. A useful feature of the ITS2 Database is the web interface that enables taxon sampling, secondary structure prediction, sequence–structure based alignment, and tree reconstruction.
Databases for fungal genetic markers are also available. AFTOL (Assembling the Fungal Tree of Life) [180] significantly contributed to the understanding of the evolution of the Kingdom Fungi, making sequence data, alignments, and other types of fungal data available to the scientific community. Among its contributions is a list of primers of interest for fungal taxonomic and phylogenomic studies. SPPADBASE [181] is an online searchable database of primer sets for the detection and identification of plant pathogenic fungi. It includes over 570 primer sets for more than 200 species of phytopathogenic fungi. DFVF [182] is a database of fungal virulence factors, and includes information on 2,058 genes of fungal pathogens.
The Barcode of Life Data Systems (BOLD SYSTEMS) [183] includes a database dedicated to DNA barcode data, comprising barcodes for Animals, Plants, Protists, and Fungi. It currently contains 14,992 fungi-specific barcodes and a comprehensive registry of primers used in the generation of barcode sequences, maintained by users.
Figure 2 shows a detailed analysis of the proportion of sequences from different phyla of Fungi in some of the above-mentioned databases. Ascomycota and Basidiomycota (Dikarya) are the most represented in all databases, and together they constitute more than 60% of the sequences publicly available. This high representation certainly reflects the fact that these are the largest fungal phyla known to date under the light of the methods used [150,184]. However, as pointed out by Bass and Richards [42], this scenario is likely to change in the future, when we learn more about some of the newly described higher level taxa, such as Cryptomycota [185,186] and Archaeorhizomycetes [187-189].
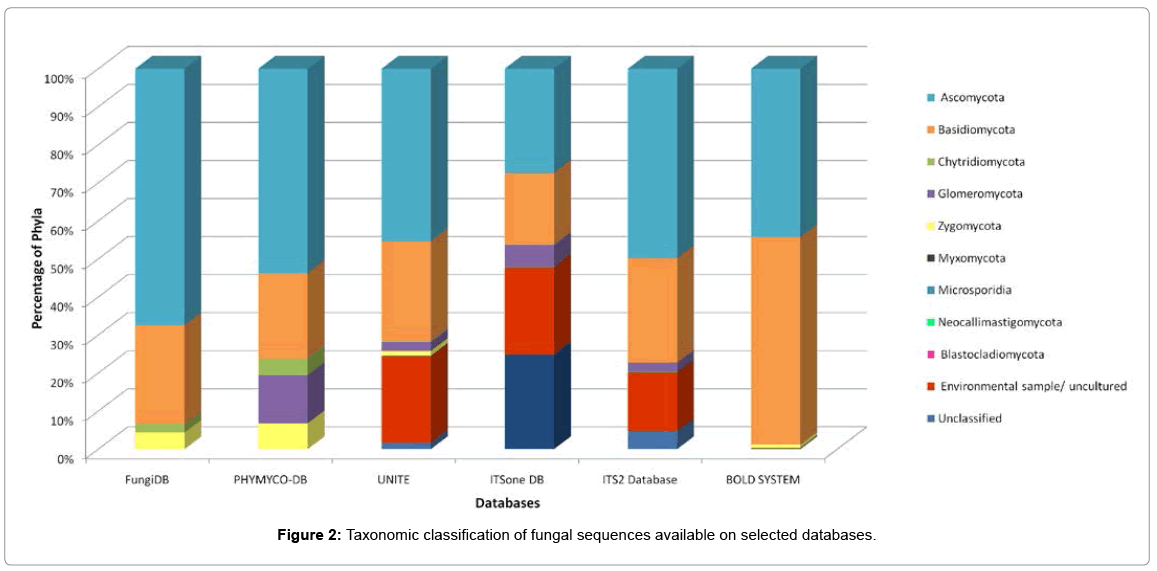
Figure 2: Taxonomic classification of fungal sequences available on selected databases.
Noteworthy is the fact that some databases, such as UNITE, ITSone DB and ITS2, contain a high percentage of sequences annotated as environmental sample / uncultured. This is clear evidence of the need for more studies focusing on fungal genomics and metagenomics in order to increase the taxonomic knowledge about this kingdom, and improve classification of environmental sequences.
The Joint Genome Institute (JGI, DOE/US) has made a substantial contribution to genomic and metagenomic research by creating and maintaining important and fully integrated databases, such as IMG/M and IMG/HMP M, among other contributions. Figure 3 shows the diversity of sequences assigned to known fungal phyla in IMG/M database. JGI is also the official sequencing center for the 1000 Fungal Genomes Project (F1000), which derived from the AFTOL Project and is aimed at filling in gaps in AFTOL. This effort will generate useful reference information for research on plant-microbe interactions, microbial emission, and capture of greenhouse gases, as well as environmental metagenomic sequencing that can be useful in future comparative studies.
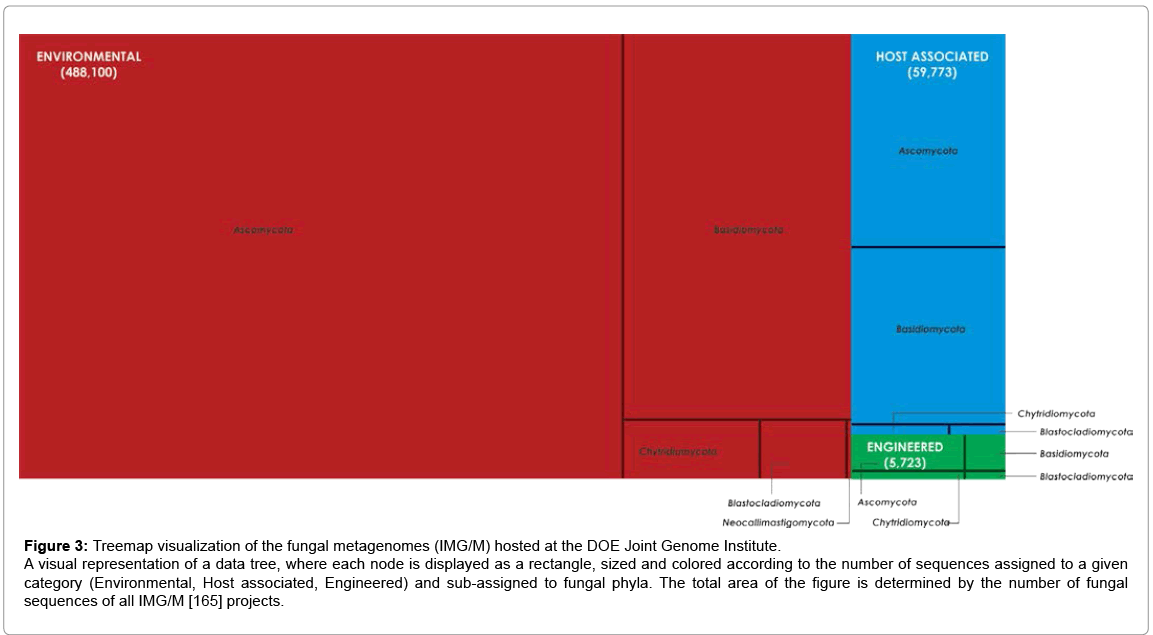
Figure 3: Treemap visualization of the fungal metagenomes (IMG/M) hosted at the DOE Joint Genome Institute.
A visual representation of a data tree, where each node is displayed as a rectangle, sized and colored according to the number of sequences assigned to a given category (Environmental, Host associated, Engineered) and sub-assigned to fungal phyla. The total area of the figure is determined by the number of fungal sequences of all IMG/M [165] projects.
NGS catalysed the research on microbial community genomics, and paved the way for scientists to build fundamental knowledge on fungal communities in the environment over the past decade. The possibility of generating large datasets of sequences from both culturable and unculturable microorganisms has allowed for the study of mixed samples in a less biased way, which is a prerequisite for ecological and host-microorganism association studies.
Significant advances have been made in sampling methods, in order to enrich the biomass with eukaryotic cells and thus allow for a higher coverage of these genomes. Improvements have also been made in computational methods in recent years, for example in bioinformatic algorithms and databases. New approaches and methods have been proposed. Comparative metagenomics, for instance, is a field of research under development, and it is challenging due to the complex nature of microbial communities, together with the fact that different sequencing methods have been used to generate metagenomic datasets, producing reads that vary widely both in total number and average length [135]. Quantitative metagenomics is still in its infancy, and the new methods developed rely on a normalization of data based on the average genome size of the organisms sampled [135].
In spite of these methodological advances, the metagenomic approach still has inherent limitations. For instance, it cannot distinguish live from dead or active from inactive microbial cells. Furthermore, the computational assembly of metagenomic data is at the risk of chimera generation, leading to mistakes in microbial diversity interpretation. Alternative splicing and single nucleotide polymorphisms (SNPs) take eukaryotic diversity studies to a higher level of complexity when compared to prokaryotic genomics, mainly because of their potential to generate phenotypic variation.
Our research group has used a targeted metagenomic approach and massively parallel sequencing to investigate the diversity of fungi associated with decaying wood in a tropical forest [manuscript in preparation]. With a growing numbers of environmental samples being collected and sequenced worldwide, we might be able scratch deeper into the true variation of this amazingly diverse eukaryotic group.
Further advances in data generation, especially longer reads, and analysis methods may minimize errors and misinterpretations, making cross-analysis of metagenomes more feasible. Certainly, the integration of metagenomic, metatranscriptomic and metaproteomic data in open access databases will positively affect microbial ecology research and fungal diversity and ecology studies, in particular.