Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2016) Volume 5, Issue 5
Keywords: B-Cell; Multiple sclerosis; Secondary progressive multiple sclerosis
As physicians and neuroscientists, we would like to prevent the development of increasing disability in multiple sclerosis (MS), which puts a great burden on the patient and caregivers [1]. In 1996, the US National Multiple Sclerosis Society (NMSS) Advisory Committee on Clinical Trials in Multiple Sclerosis defined the clinical subtypes of multiple sclerosis (MS) [1]. The Committee provided standardized definitions for four MS clinical courses: relapsing-remitting (RR), secondary progressive (SP), primary progressive (PP), and progressive relapsing (PR) [1]. Either relapsing or progressive disease can be characterized by severity of signs and symptoms, frequency of relapses, rate of worsening, residual disability, and impairment [1]. Primary progressive multiple sclerosis (PPMS) accounts for 10%-15% of the entire MS population [2]. Two thirds of all patients with MS present relapsing remitting multiple sclerosis (RRMS) [3] characterized by clinically evident bouts of disease activity. Disability also accrues as disease progresses. As showed in Figure 1, the relation between inflammation and neuroaxonal loss during the progression of MS is particular: during the early phase of MS inflammation grow. In the same period, neuroaxonal loss is at low level. When the disease turn in RRMS inflammation decrease while neuroaxonal loss increases its level. In PPMS phase, the grade of inflammation becomes low while the neuroaxonal loss became preponderant. Cognitive, visual, and other clinical changes could provide clinical evidence for disease activity [1]. Once patients have transitioned from RRMS to secondary progressive multiple sclerosis (SPMS), the burden of disability becomes greater. Moreover, it is well known how disability relentlessly progresses from the start. Degree of recovery from an acute relapse was considered to be, in itself, not useful for determining or modifying MS phenotypes, but is instead a contributor to disease worsening over time [1]. We suggest using the term worsening in place of progressing especially for patients with relapsing forms of disease, reserving the term progression only for those in the progressive phase of MS, independent of relapse activity [1].
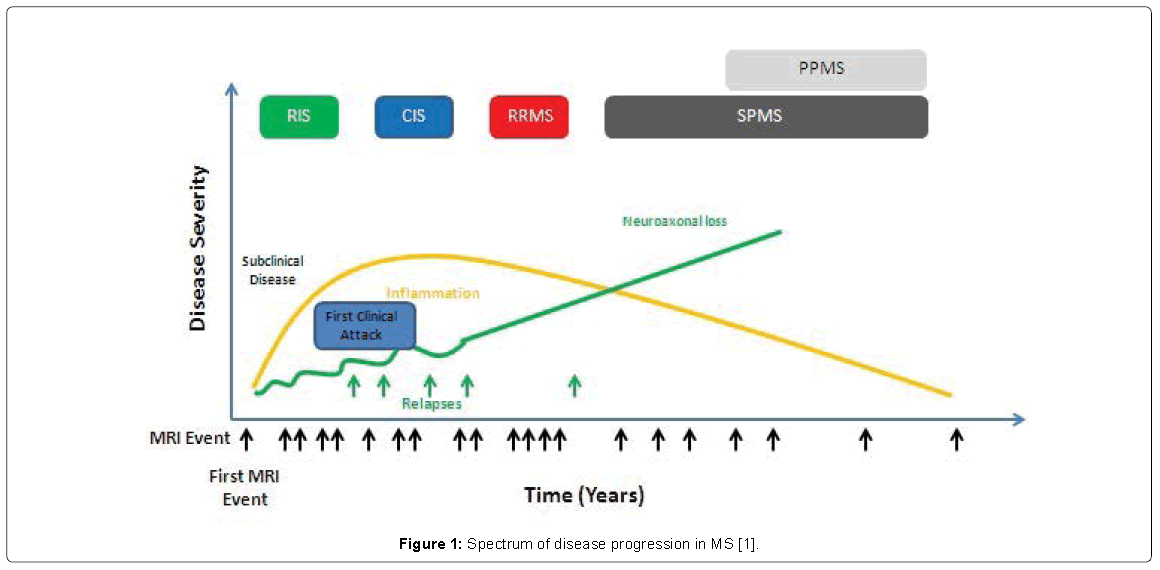
Figure 1: Spectrum of disease progression in MS [1].
Currently, inflammation and neurodegenerative component are the main substrate thought to be involved in the pathogenesis of diseases; indeed [4], during the earlier stages of RRMS, the disease is fully driven by strong inflammatory responses orchestrated by autoreactive T or B cells. As the disease evolves and as the progressive features dominate, the neurodegenerative mechanisms govern the evolving deficits and disability However, in many instances, a mix of these processes is probably occurring (Figure 2) [5,6]. Tissue changes can be induced by several different mechanisms, directly related to the inflammatory process via T-cell and B-cell mediated inflammation or dependent on microglial activation leading to mitochondrial injury and oxidative burst [7]. Secondary complications include energy deficiency [7]. New MS lesions begin with perivenular cuffing by inflammatory mononuclear cells, predominantly T cells and macrophages, which infiltrate the surrounding white matter [8]. At sites of inflammation, the blood-brain barrier (BBB) is disrupted, but unlike vasculitis, the vessel wall is preserved [9]. Involvement of the humoral immune system is also evident; small numbers of B lymphocytes also infiltrate the nervous system, and myelin-specific autoantibodies are present on degenerating myelin sheaths [10]. As lesions evolve, there is prominent astrocytic proliferation (gliosis) [11]. Surviving oligodendrocytes or those that differentiate from precursor cells can partially remyelinate the surviving naked axons, producing so-called shadow plaques [10]. In many lesions, oligodendrocyte precursor cells are present in large numbers but fail to differentiate and remyelinate [8-11]. Over time, ectopic lymphocyte follicles appear in perivascular and perimeningeal regions, consisting of aggregates of T and B cells and resembling secondary lymphoid structures [12]. Although relative sparing of axons is typical of MS, partial or total axonal destruction can also occur, especially within highly inflammatory lesions [4-9]. Thus, MS is not solely a disease of myelin, and neuronal pathology is increasingly recognized as a major contributor to irreversible neurologic disability [1-3]. Inflammation and plaque formation are present in the cerebral cortex, and significant axon loss indicating death of neurons is widespread, especially in advanced cases [4]. Axonal damage occurs in every newly formed MS lesion, and cumulative axonal loss is considered to be the major cause of progressive and irreversible neurologic disability in MS [5]. As many as 70% of axons are lost from the lateral corticospinal (e.g., motor) tracts in patients with advanced paraparesis from MS, and longitudinal MRI studies suggest there is progressive axonal loss over time within established, inactive lesions [1,2]. Knowledge of the mechanisms responsible for axonal injury is incomplete and, despite the fact that axonal transactions are most conspicuous in acute inflammatory lesions, it is still unclear whether demyelination is a prerequisite for axonal injury in MS [9]. Demyelination can result in reduced trophic support for axons, redistribution of ion channels, and destabilization of action potential membrane potentials [10]. Axons can adapt initially to these injuries; with time, distal and retrograde degeneration often occurs [9,10]. Therefore, promotion of remyelination and preservation of oligodendrocytes early in the disease course remain important therapeutic goals in MS [12]. Some evidence suggests that axonal damage is mediated directly by resident and invading inflammatory cells and their toxic products, in particular by microglia, macrophages, and CD8 T lymphocytes [7]. Activated microglia are particularly likely to cause axonal injury through the release of NO and oxygen radicals and via glutamate, which is toxic to oligodendrocytes and neurons [7,8]. Interestingly, NMDA (glutamate) receptors are expressed on naked axon membranes that have undergone demyelination, perhaps providing a mechanism for glutamate-mediated calcium entry and cell death [8].
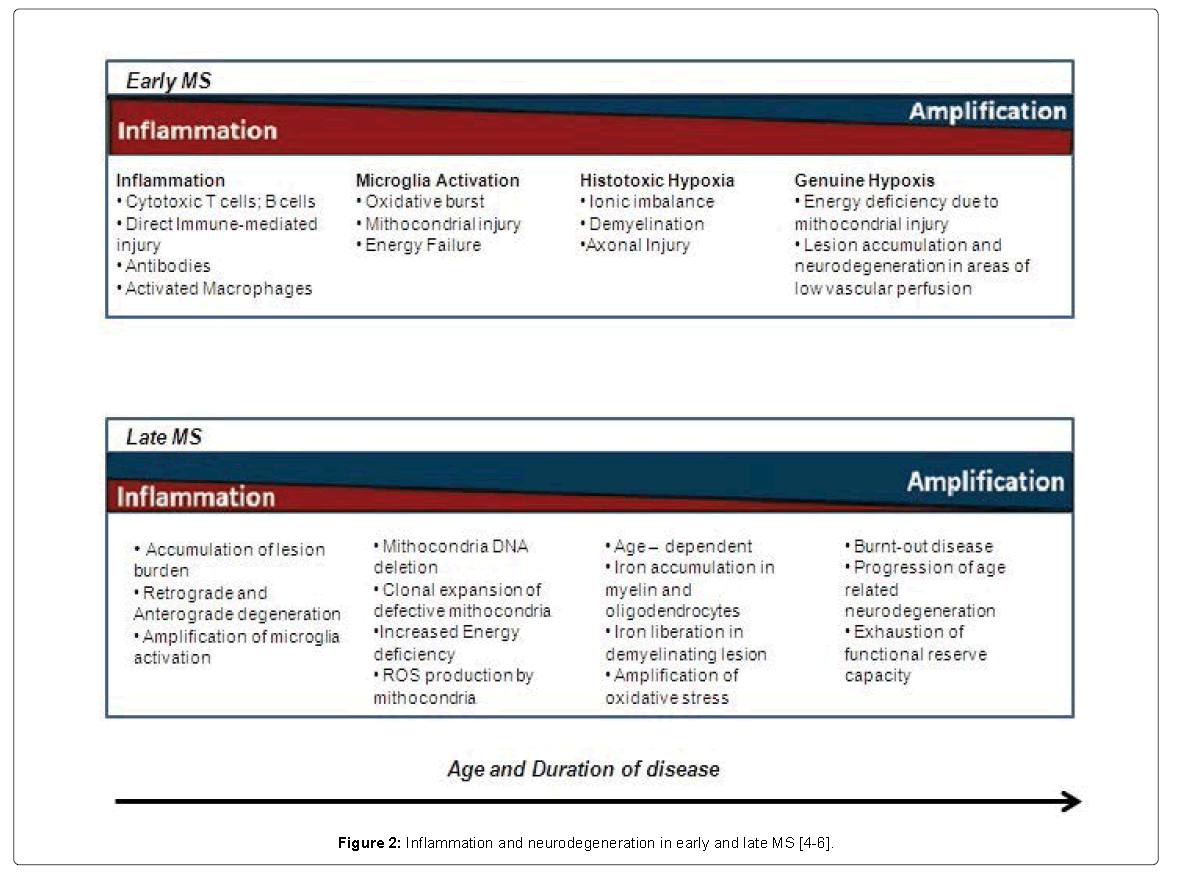
Figure 2: Inflammation and neurodegeneration in early and late MS [4-6].
In RRMS, T and B cells in the periphery play a very important role. In the progressive forms of MS, there may be a very decisive role played by innate immune system cells [7]. In the brain, these would be microglial cells. The history of MS has been focused primarily on T cells for some very good reasons [4]. First, the experimental model of autoimmune encephalomyelitis (EAE), particularly in mice, involves cell-directed immunopathology in the brain that simulates the acute activity with associated demyelination occurring in the brains of MS patients [5]. Nonetheless, this model misses to explain the progressive disease. It is even a flawed model for MS in general. The one thing about the model is that we can adoptively transfer the disease from an affected mouse to an unaffected mouse by using pure populations of activated T cells. That has probably driven much of the therapy focused on controlling T-cell subsets in MS. Nevertheless, we have known for many decades that B cells might have some role in MS because one of the characteristic biomarkers of the disease is oligoclonal bands in the cerebrospinal fluid (CSF) [6]. This finding implicates B cells as making immunoglobulins within the central nervous system (CNS). While we have never been able to determine what those immunoglobulins are doing there or how they might be specifically driving the disease, they are such a characteristic phenomenon of both RRMS and PPMS that one wonders why B cells might be a novel appealing target in MS treatment.
Indeed, leptomeningeal inflammation from secluded compartments of B-cell aggregates might contribute to the pathogenesis that leads to continuous disease progression [7].
The traditional view is that B cells are the source of autoantibodies crucial to the demyelination process [8]. More recently, B cells have an emerging role as antigen-presenting cells that instruct T cells to begin the immunoinflammatory cascade [9]. B cells are also thought to be builders of the local immune system, for example, in the progressive stages of MS (Figure 3) [10,11].
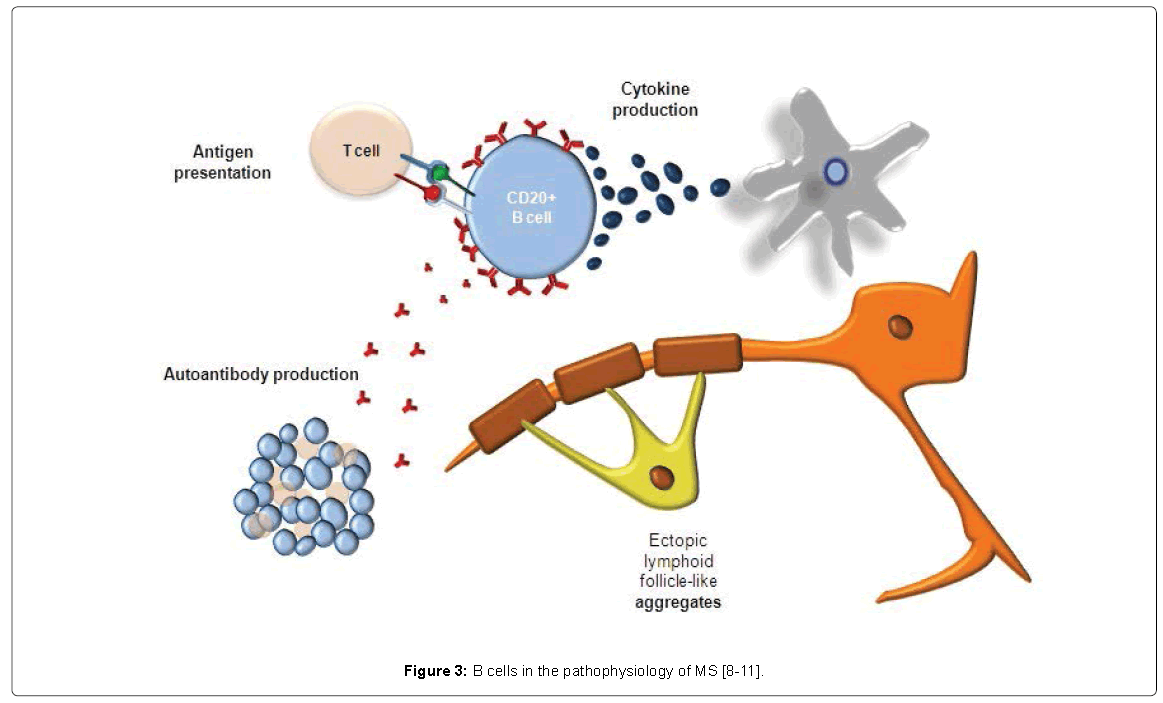
Figure 3: B cells in the pathophysiology of MS [8-11].
Moreover, in RRMS and PPMS, B cells play a very important role in the central nervous system (CNS)-occurring immune responses, forming the so-called compartmentalized inflammation and folliclelike structures. Nonetheless, this role still needs to be further explored [4]. Since inflammation occurs behind a closed BBB and B cells play a crucial role, anti-inflammatory drugs that act in the periphery have difficulty passing either a closed or a repaired BBB; furthermore, intrathecal anti-inflammatory therapy might be able to circumvent this. This might be the reason why BBB damage does not appear anymore a good measure of ongoing inflammatory disease activity [4].
Anti-CD20 Therapy in MS: Mechanisms of Action and Clinical Experience
Recent years have brought an emerging interest in the arena of B cell-targeted therapies Anti-B-cell therapy -- depletion or silencing of B cells-could conceptually work via several distinct mechanisms [12]. The background of effectiveness of immune mediated approach is the interaction with antigen presentation by CD20-positive B cells and consequent immunosuppression of T cells. Nevertheless, also the depletion of CD20 positive T cells corroborate this effect [13,14], in combination with anti-inflammatory effect by diminishing of proinflammatory cytokines produced by B cells [15]. It is also been shown that reduction of antibody production (in blood) with long-term treatment is related to a slight decline of IgM concentration, together with a largely unaltered IgG subclass [13]. On the other hand, T cells and CXCL13 are reduced in CSF [16]. On this basis B-Cell Depletion with Rituximab in RRMS: phase 2 HERMES trial, was conducted on 104 patients, randomly assigned 2:1; 69 patients received 1000 mg of intravenous Rituximab, and 35 patients received placebo; this trial showed that Rituximab really silenced the disease, which was the basis for using anti-CD20 in RRMS (Figure 4) [12].
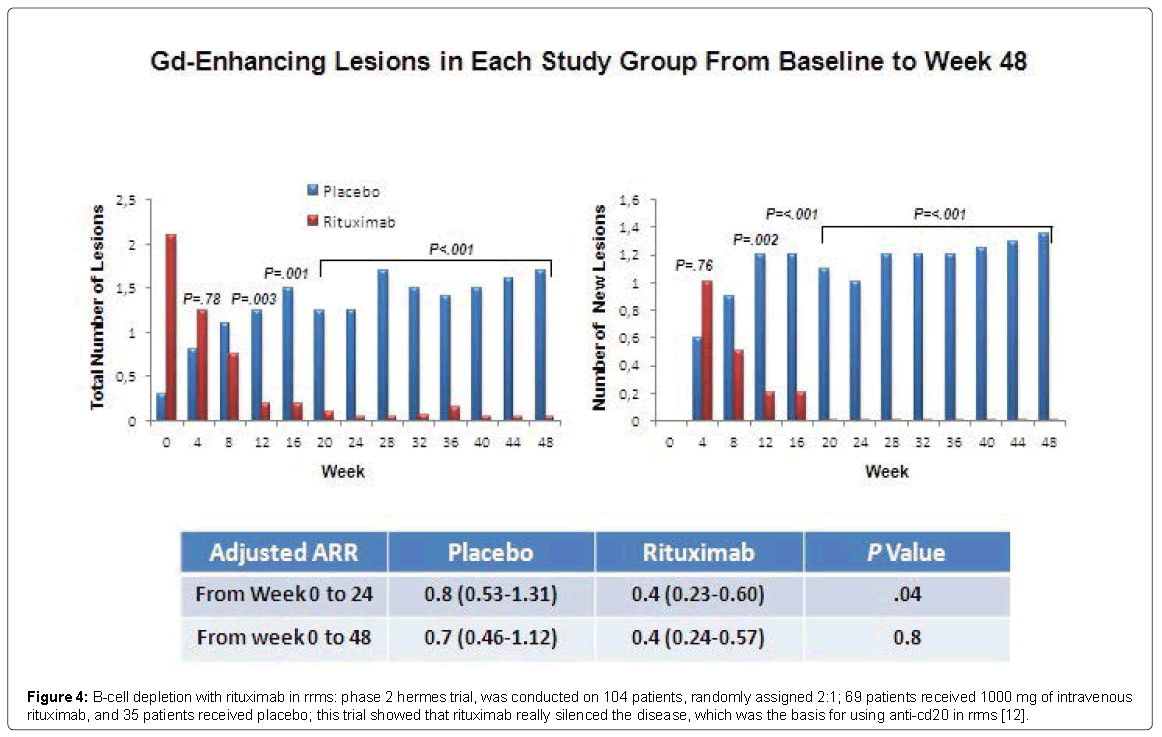
Figure 4: B-cell depletion with rituximab in rrms: phase 2 hermes trial, was conducted on 104 patients, randomly assigned 2:1; 69 patients received 1000 mg of intravenous rituximab, and 35 patients received placebo; this trial showed that rituximab really silenced the disease, which was the basis for using anti-cd20 in rrms [12].
Emerging data on B cell-targeting therapies have focused on a new anti-CD20 monoclonal antibody, ocrelizumab [17]. There are differences between rituximab and ocrelizumab from a molecular standpoint, as shown in Figure 5.
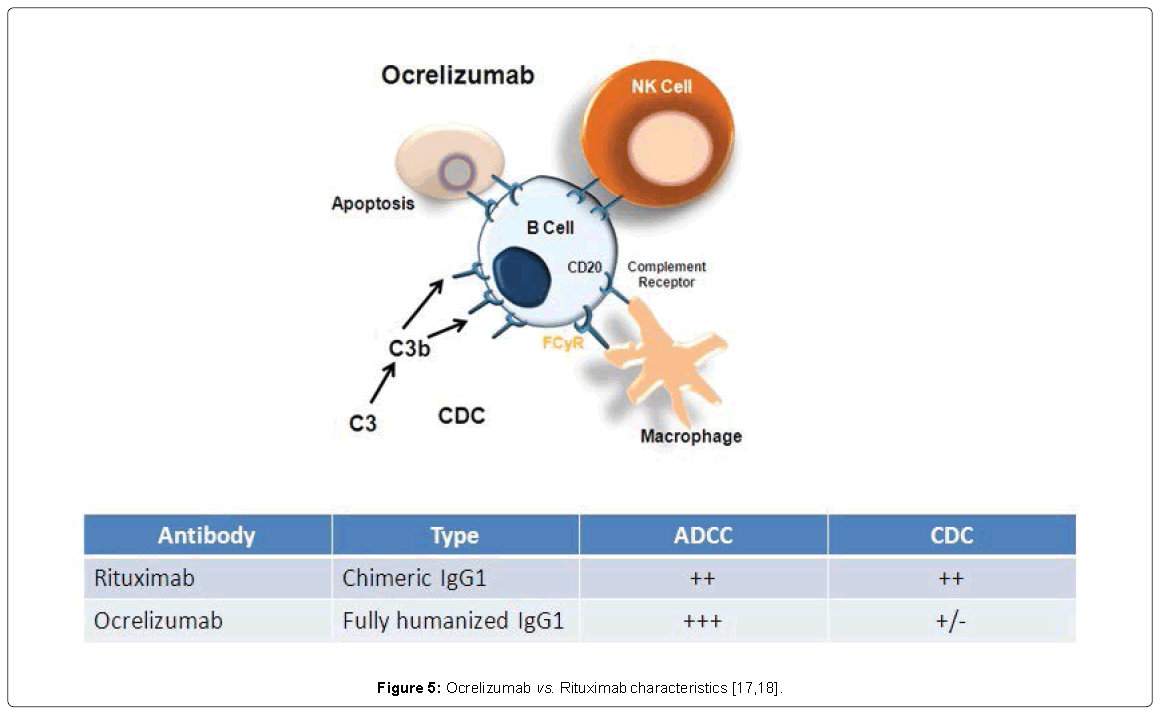
Figure 5: Ocrelizumab vs. Rituximab characteristics [17,18].
In addition: the epitopes recognized on CD20 by Rituximab and ocrelizumab are distinct but overlapping. The stronger antibodydependent cellular cytotoxicity with ocrelizumab might translate to increased therapeutic efficacy. The reduced complement dependent cytotoxicity explains the lower frequency of infusion-related reactions (IRRs) [18]. Ocrelizumab has a modified Fc portion that potentially reduces AEs related to complement activation (compared with rituximab). Additionally, higher affinity for FcgRIIIA, including low affinity it has been developed. Overall, improved potency and potentially improved efficacy/safety profile has emerged [18]. Ocrelizumab has been investigated in two Identical Phase 3 Studies Evaluating in RRMS [19]. Data were recently released from the OPERA studies, which investigated the use of ocrelizumab in RRMS. These were 2 identical studies with ocrelizumab vs subcutaneous high-dose Interferon (IFN) β, 3 times weekly. In 2 years, ocrelizumab showed great results not only clinically, with annualized relapse rate (ARR) and confirmed disability progression (CDP), but also with MRI and brain atrophy [19]. The primary endpoint was significant reduction in ARR compared with IFNβ-1a and the secondary endpoints concerned significant reduction in CDP in the prespecified pooled analysis of OPERA OPERA II, and OPERA II, with reduction in MRI Lesions [19]. There have been many recent publications confirming a high correlation between brain atrophy and the development of disability, underlying the complementary predictive value of atrophy and lesion volumes for predicting long-term disability in MS [18,19]. Nonetheless, the reduction in brain volume loss with ocrelizumab was closer to what is seen in normal healthy controls [19]. What was predictable from the Rituximab experience, even if still concerning is IRRs, which can be decreased by pretreatment with steroids and anti-inflammatory drugs.
There was a little bit higher proportion of common infections, but nothing opportunistic, and of malignancies [19]. In light of these data, B cells are no longer strictly a diagnostic measure (oligoclonal bands- OCBs) in the CSF. Trial data seems to force to rethink the role of the B cell in MS. It is clear that B cells play a significant and pathogenic role because B-cell depletion has a large effect on multiple measure (ARR, MRI) (Figure 6) [20].
Figure 6: B cells are no longer strictly a diagnostic measure (oligoclonal bands – OCBs) in the CSF. Trial data seems to force to rethink the role of the B cell in MS. It is clear that B cells play a significant and pathogenic role because B-cell depletion has a large effect on multiple measures (i.e.MRI) [20].
Other B cell-targeting therapies have failed in MS, including atacicept. Atacicept is a fusion protein that acts as a receptor for important survivor B-cell survival factors including BAFF and APRIL. This trial showed that not only patients did not receive any benefit from atacicept, but also they actually did worse on it [21]. Immunologic data for the reason behind these results are not available, but there is a theory: there are different populations of B cells; one drives proinflammatory activity, and the other regulates and counteracts overactive inflammation. It is possible that atacicept broadly targeted the B-cell population, including "regulatory" B cells, and deprived them of their survival factors. As a result, the regulatory influence of these cells was no longer exerted, and pathogenic activities prevailed. In light of such evidence is mandatory to validate any B-cell directed approach [21].
The results with fingolimod were particularly surprising [22]. It was considered an attractive candidate because of data showing that it acts both on the peripheral immune system and in the CNS [23-25].
A subgroup analysis of the Olimpus study showed that patients under 51 years of age, with at least one gadolinium (Gd)-enhancing lesions at baseline, fared much better than other patients [26] did. However, the number of patients was small and along with disease progression, the pathogenesis seems to be due to aging.
The older patients do not profit as much from this therapy because they have this additive pathology. Instead, the younger patients with active lesions on MRI may profit.
MD1003 (High-Dose Biotin) incorporate a different mechanism, reversing energy deficiency in demyelinated axons; biotin acts as a cofactor for enzymes expressed in astrocytes and neurons that are central to aerobic energy production. Increased availability of intraneuronal pool of ATP leads to reduced demyelinated neural dysfunction and adverse effects of hypoxia. Moreover activates myelination formation in oligodendrocytes, when the disease is active, there is inflammation, and so anti-inflammatory drugs can be used acting as cofactor for the rate-limiting enzymes in fatty acid biosynthesis required for myelin synthesis [4]. Increased availability of these enzymes leads to increased fatty acid synthesis required for myelin repair [27]. While the number of patients in this trial was small, the findings are still intriguing [28]. Interesting but very cautious sign of hope. Further exploration is needed with replication of these data in a larger controlled trial setting [29].
The ORATORIO study enrolled more than 700 patients, who were randomly assigned to either placebo or ocrelizumab. The duration of the study was 120 weeks to be able to determine the 24-week CDP [30]. The average age was higher than in RRMS trials, around 44 years. It is in the range where OLYMPUS showed positive results. Around 25% of patients were also positive for Gd-enhancing lesions. The reduction in the risk for CDP is very encouraging. Because there is a connection between brain atrophy and disability progression, it is reassuring to see that ocrelizumab also had an effect on brain volume loss in PPMS within 120 weeks. There were also increased IRRs in this trial, as well as slightly increased common infections. There were also a slightly increased number of malignancies. This is important because of the mismatch in the occurrence between ocrelizumab and placebo. These are relatively small numbers, but we have to be cautious. Additional data are needed to see whether this signal will persist and stand out against the background prevalence of malignancies in this patient or age group.
The Role of B cells in the Pathogenesis of PPMS seems to be Similar to that in RRMS.
However, there could be some differences in the PPMS population. It is still a hypothesis that anti-CD20 therapy has an impact on compartmentalized inflammation associated with progression. It is possible that the anti-CD20 antibodies could go through the bloodbrain barrier, target the B-cell follicles, and have a therapeutic effect in the CNS [12-16]. The patient population in ORATORIO was relatively young compared with other big trials conducted in PPMS, and a significant proportion of patients also had inflammatory activity on baseline MRI. A recently presented post hoc analysis of the ORATORIO data suggests that there was no difference in time to onset of 12- or 24-week CDP in patients who had Gd-enhancing lesions at baseline, compared with those who did not. Many open questions remain, but these data provide momentum and hope for patients with PPMS who have been left out from the advances in therapies developed over the years for RRMS [31].
Potential future roles for B cell-targeted therapies in MS, B celldirected therapies are effective in both RRMS and PPMS. If they are effective in clinically isolated syndrome and SPMS, there would be a therapeutic approach that covers the entire "spectrum of MS". Physicians may adopt ocrelizumab for the management of SPMS, which constitutes a far larger proportion of the progressive population than PPMS. Ideally, B cell-directed therapies would be given at the beginning of PPMS, as soon as it is diagnosed or at least within 1 year. Based on the data, there is no reason to wait longer to start therapy.
Ofatumumab is a fully human antibody, which targets epitope distinct from rituximab, with similar ADCC to rituximab but stronger CDC, even with low CD20 density. Moreover, ofatumumab it is characterized by a more potent and prolonged B-cell depletion. A phase 2 studies it has been completed and phase 3 results are expected. Anti-CD19 antibodies are currently being developed for clinical trials. The most advanced compound is MEDI-551, an affinity-optimized and afucosylated humanized IgG1 kappa, with effect on B-cell depletion primarily dependent on ADCC [18,32,33]. There is a need to find reliable markers of B-cell depletion, when Rituximab was being investigated; the persistence of B-cell depletion was gauged by measuring CD19 cells in the periphery. There was no reliable correlation. Therefore, the decision to reintroduce Rituximab was not based on peripheral blood CD19- positive cells but on the patient's clinical picture. Data in experimental models suggest that there is a disproportionate effect of B-cell depletion in the periphery vs tissues. It is currently not known how frequently or for how long a powerful B cell-depleting agent can be administered to patients with MS without potentially encountering risks [13,14]. Moreover, robust evidence regarding the long-term use of B-cell therapy in MS is scanty. Nevertheless, a long-term safety and efficacy study it has been designed. Data were retrieved from the Swedish MS registry and through medical chart review for all patients ever treated with rituximab. Within 753 MS patients treated with B-cell depletion therapy, fewer than 15 severe adverse events (grade 3 or more) were detected, including generalized arthralgia, palmoplantar pustolosis, bilateral noninfectious pulmonary infiltrates, and infusion related reactions. Preliminary analysis indicate a 1-year drug survival >90%. Rituximab showed a high degree of drug survival and a good tolerability profile with few treatment interruptions due to inadequate treatment response [34]. Nonetheless, data will have to be carefully collected, most likely in the context of a registry, to reassure that the good emerging safety profile of ocrelizumab from the pivotal trial will hold up [35].
None of the authors has any competing interests.