Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 1
Alzheimer’s disease is a progressive neurodegenerative disorder characterized by deposition of amyloid plaques composed of aggr e- gated amyloid beta plaques, and neurofibrillary tangles composed of hyperphosphorylated tau that leads to synaptic defects resu lting in neuritic dystrophy and neuronal death. Missense mutations in amyloid precursor protein (APP), PS-1 (presenilin-1 situated on chromosome 14), PS-2 (presenilin-2 situated on chromosome 1) genes alter the proteolysis of APP and increase the generation of Aâ42 (amyloid â-42). The accumulation of Aâ42 as diffuse plaques triggers the inflammatory responses in the form of microglial activ ation and release of cytokines. In addition, perturbation of equilibrium between kinases and phosphatases results in hyperphosporylat ion of tau protein. These events culminate in neuronal degeneration and neuronal loss.
In the present study, we extracted huge amounts of data from various biological databases available online. It is found that th ere are 74 genes that may cause Alzheimer’s disease .We evaluated the role of 74 proteins that are likely to be involved in Alzheimer’s di sease by employing multiple sequence alignment using ClustalW tool and constructed a Phylogenetic tree using functional protein sequence s extracted from NCBI. Phylogenetic tree was constructed using Neighbour – Joining Algorithm in Bioinformatics approach. The results of this study suggest that PS-1, PS-2, and APP have a dominant role in the pathogenesis of Alzheimer’s disease. The pre sent study raises the possibility that genetic components are more important in Alzheimer’s disease compared to environmental, metab olic, and age related factors.
Keywords: Alzheimer’s disease; Free radicals; Presenilin; Acetylcholine; Phylogenetic trees; Amyloid precursor protein
Over the past 25 years, it has become clear that the proteins forming the deposits are central to the disease process. Amyloid-ß and tau make up the plaques and tangles of Alzheimer’s disease(AD), where these normally soluble proteins assemble into amyloid-like filaments (Michel and Maria, 2006) . Recently Ballatore et al., (2007) summarized the most recent advances in the mechanisms of tau-mediated neurodegeneration to forge an integrated concept of those tau-linked disease processes that drive the onset and progression of AD and related tauopathies. New evidence indicates that tau may mediate neurotoxicity by altering the organization and dynamics of the actin cytoskeleton (Gallo, 2007). Amyloid formation is a nucleation-dependent process that is accelerated dramatically in vivo and in vitro upon addition of appropriate fibril seeds (Alexander et al., 2006) AD is a progressive neurodegenerative disorder characterized by amyloid plaques composed of aggregated amyloid beta plaques, neurofibrillary tangles (NFT) composed of hyperphosphorylated tau and synaptic defects resulting in neuritic dystrophy and neuronal death (Hutton and McGowan, 2004). A growing body of evidence implicates cholesterol and cholesterol-rich membrane microdomains in amyloidogenic processing of amyloid precursor protein (APP).Cheng et al., (2007) reviewed the recent findings regarding the association of BACE1,γ-secretase and APP in lipid rafts, and discuss potential therapeutic strategies for AD that are based on knowledge gleaned from the membrane environment that fosters APP processing.
Missense mutations in amyloid precursor protein (APP), presenillin-1 (PS-1) (chromosome 14), presenillin-2 (PS-2) (chromosome 1) genes alter the proteolysis of APP and increase the generation of Aβ42 (amyloid β 42) .Genetic studies have led to the identification of three genes in which mutations can cause AD: the ß-amyloid precursor protein gene located on chromosome 21, presenilin 1 (PS1) located on chromosome 14 and presenilin 2 (PS2) located on chromosome 1 (Hanuman et al., 2007). The accumulation of Aβ42 as diffuse plaques triggers the inflammatory responses due to microglial activation and release of pro-inflammatory cytokines. In addition, perturbations in the equilibrium between kinases and phosphatases resulting in hyperphosphorylation of tau protein that results in neuronal degeneration and neuronal loss (Selkoe, 2001). The microtubuleassociated protein tau is also involved in the disease, but it is unclear whether treatments aimed at tau could block Aß-induced cognitive impairments (Erik et al., 2007).
Several other genes that are considered to increase susceptibility for AD include: apolipoprotein E (ApoE 4) variant (Poierier et al., 1995), 2-macroglobulin (Blacker et al., 1998), the K-variant of butyryl-cholinesterase (Sridhar et al., 2006), and several mitochondrial genes (Law et al., 2001). Other factors that are believed to play a role in the aetiopathogenesis of AD include: brain metabolic abnormalities, environmental factors, and age related decrease in neuronal membrane fluidity that could also produce neuronal death, in all probability, by increasing the formation of amyloid beta plaques and hyperphosphorylation of tau protein (Iqbal et al., 2005).
Mutations in presenilins leads to dominant inheritance of Familial Alzheimer’s disease (FAD). These mutations are known to alter the cleavage of γ-secretase of the amyloid precursor protein, resulting in the increased ratio of Aβ42/ Aβ40 and accelerated amyloid plaque pathology in transgenic mouse models (Wang et al., 2006). Proteolytic processing of APP by β-secretase, γ - secretase, and caspases generates A-beta peptide and carboxylterminal fragments (CTF) of APP, which have been implicated in the pathogenesis of Alzheimer’s disease (Selkoe, 1999). Missense mutations in the gene encoding APP, as well as those in the genes encoding PS-1 and PS-2, share the common feature of altering the γ-secretase cleavage of APP to increase the production of the amyloidogenic Aβ42, a primary component of amyloid plaques in both familial and sporadic AD.
In the present study, we focused on the genes or proteins that are believed to have a major role in the pathogenesis of Alzheimer’s disease using bioinformatics tools.
We collected 74 known proteins that are believed to be involved in the pathogenesis of Alzheimer’s disease (Table 1). The functional protein sequences in FASTA format for these proteins are collected from NCBI (National Center for Biotechnology Information, (http\\www.ncbi.nih.nlm.gov). These sequences are given to ClustalW (http\\www.ebi.ac.uk\clustalw) for the Multiple Sequence Alignment, which calculates the best match for the selected sequences, and lines them up so that the identities, similarities and differences can be seen. Based on these results, the scores table and phylogenetic tree that shows the distance between the protein sequences was constructed. The proteins with minimum distance are presenillin-1 (PS-1), presenillin-2 (PS-2) and amyloid precursor protein (APP).
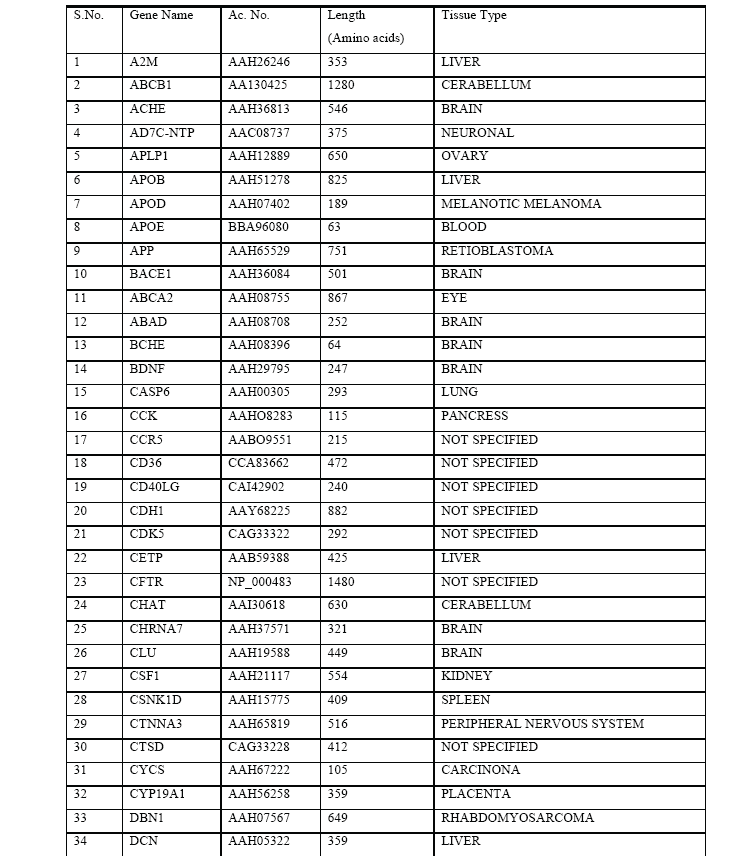
The bioinformatics analysis revealed three important proteins out of 74 proteins that are key pathological proteins in the evolution of Alzheimer’s disease. The present bioinformatics study revealed that the proteins: presenilin-1 (PS-1), presenilin-2 (PS-2), and amyloid precursor protein (APP) play a significant role in the pathogenesis of Alzheimer’s disease (Figure 1).

Figure 1: The phylogenetic tree that was constructed based on the alignment score of all the protein sequences involved in Alzheimer’s disease. A high degree of homology was noted between presenilin1, presenilin 2, Amyloid beta (A4) precursor protein.
Factors that seem to influence the initiation and progression and thus, have a role in the pathophysiology of AD are: i) Aβ42/ Aβ40 ratio and oligomers of these peptides; ii) oxidative stress; iii) proinflammatory cytokines produced by activated glial cells, iv) alterations in cholesterol homeostasis, and v) alterations in cholinergic nervous system (Rojo et al., 2006).
Amyloid Beta Peptide and Alzheimer’s Disease
Familial Alzheimer’s disease (FAD) is associated with mutations in APP, PS-1, and PS-2.
These substances, along with their normal counterparts, undergo proteolytic processing in the endoplasmic reticulum (ER). The mutated compounds, apart from increasing the ratio of Aβ42 to Aβ40, may down-regulate the calcium buffering activity of the ER. Decrease in the ER calcium pool would cause compensatory increases in other calcium pools, particularly in mitochondria. Increase in mitochondrial calcium levels are associated with enhanced formation of superoxide radical formation, and hence damage to the neurons and their senility (Harman, 2002).
Presenilins act as catalytic subunit of gamma secretase. Presenilins, the causative molecules of FAD, are transmembrane proteins localized predominantly in the ER and Golgi apparatus. Presenillins are thought to be involved in intramembrane proteolysis mediated by their gamma secretase activities. In addition, presenilins interact with FKBP38 (human FK506-binding protein 38) and form macromolecular complexes together with antiapoptotic Bcl-2, thus it may regulate the apoptotic cell death (Wang et al., 2005).
Presenilins and their interacting proteins play a major role in the generation of A-beta from the amyloid precursor protein (APP). Three proteins nicastrin, aph-1 and pen-2 interact with presenillins to form a large enzymatic complex known as gamma secretase that cleaves APP to generate Aβ (Verdile et al., 2007).
There are numerous proteases in the brain that could potentially participate in Aβ turnover. Aβ (amyloid-beta) degrader candidates include: cathepsin D and E, gelatinase A and B, trypsin- or chymotrypsin-like endopeptidase, aminopeptidase, neprilysin (enkephalinase), serine protease complexed with 2-macroglobulin, and insulin-degrading enzyme (Saido et al., 1998).
Genetic linkage studies have linked Alzheimer’s disease and plasma Aβ42 levels to chromosome 10q, which harbors the IDE (insulin-degrading enzyme) gene. IDE has been observed in human cerebrospinal fluid; and its activity levels and m-RNA are decreased in AD brain tissue and is associated with increased amyloid beta levels (Saido et al., 1998).
Amyloid beta is the major component of amyloid plaques characterizing Alzheimer’s disease. Amyloid beta accumulation can be affected by numerous factors including increased rates of its production and/or impaired clearance. Insulin degrading enzyme is responsible for the degradation and clearance of amyloid beta in the brain (Edland, 2004).
Several studies showed that Aβ is toxic to cultured neuronal cells and induces tau phosphorylation (Takashima et al., 1993). Tau is a microtubule-associated protein that stabilizes neuronal microtubules under normal physiological conditions, however in certain pathological conditions like Alzheimer’s disease, tau protein undergoes modifications, mainly through phosphorylation that can result in the generation of aberrant aggregates that are toxic to neurons (Avila et al., 2004). Amyloid vaccine (both passive and active immunization against amyloid) arrests and even reverses both plaque pathology and behavioral phenotypes in the transgenic animals (Morgan et al., 2000). Aβ42 fibrils can significantly accelerate neurofibrillary tangles formation in P301L mice providing further support to the hypothesis that amyloid beta could be a causative pathogenic factor. Mutations in tau give rise to nerofibrillary tangles but not plaques and mutations in APP or in the probable APP proteases give rise to both plaques and tangles indicates that amyloid pathology occurs upstream of tau pathology. Although the exact mechanism(s) by which amyloid beta causes neuronal death is not clear, there is evidence to suggest that it could enhance free radical generation and induce inflammation that could result in profound loss in the cholinergic system of brain, including dramatic loss of choline acetyltransferase level, choline uptake, and decrease in acetylcholine (ACh) level which are responsible for cognitive deficits in AD.
Oxidative Stress and Neuronal Death
One of the major age-related damaging agents are reactive oxygen species (ROS). Increased levels of ROS (also termed“oxidative stress”), produced by normal mitochondrial activity, inflammation and excess glutamate levels, are proposed to accelerate neurodegenerative processes characteristic of Alzheimer’s disease (Huber, 2006).
Amyloyd beta causes hydrogen peroxide (H2O2) accumulation in cultured hippocampal neurons (Mattson et al., 1995) that results in oxidative damage to cellular phospholipid membranes suggesting a role for lipid peroxidation in the pathogenesis of AD (Koppaka et al., 2000). The loss of membrane integrity due to Abeta-induced free-radical damage leads to cellular disfunction, such as inhibition of ion-motive ATPase, loss of calcium homeostasis, inhibition of glial cell Na+-dependent glutamate uptake system that results in NMDA receptors mediated delayed neurodegeneration, loss of protein transporter function, disruption of signaling pathways, and activation of nuclear transcription factors and apoptotic pathways.
Inflammation and Neuronal Death
Free radicals including H2O2 not only have direct neurotoxic actions but also participate in inflammation. The fact that inflammation plays a significant role in the pathobiology of Alzheimer’s disease is supported by the observation that in the early stages of the disease there is activation of microglial cells and reactive astrocytes in neuritic plaques and the appearance of inflammatory markers (Chong et al., 2001). Immune activation and/or inflammatory activity have been shown to be significantly elevated in the brains of AD patients compared with age-matched control patients (Dumery et al., 2001). Continuous neuroinflammatory processes including glial activation is seen in AD (Calingasan et al., 2002). Microglia and astrocytes would be activated, perceiving Abeta oligomers and fibrils as foreign material, since Abeta assemblies are apparently never observed during the development of brain and in the immature nervous system (Selkoe, 2001).
Beta-Amyloid fibrils have been shown to activate parallel mitogen- activated protein kinase pathways in microglia and THP1 monocytes (McDonald et al., 1998). Recently, it was reported that microglia from human AD brain exposed to Abeta produced and secreted a wide range of inflammatory mediators, including cytokines, chemokines, growth factors, complements, and reactive oxygen intermediates (Lue et al., 2001). Significant dosedependent increase in the production of prointerleukin-1, interleukin-6, tumor necrosis factor-á, monocyte chemoattractant protein-1, macrophage inflammatory peptide-1, interleukin-8, and macrophage colony-stimulating factor were observed after exposure to preaggregated Amyloid beta-42. These evidences emphasize the role of inflammation in the pathogenesis of AD.
Cholinergic System and Alzheimer’s Disease
A primary clinical symptom of Alzheimer’s dementia is the progressive deterioration in learning and memory ability. There is evidence that suggests that profound loss in the cholinergic system of brain, including dramatic loss of choline acetyltransferase level, choline uptake, and acetylcholine (ACh) level in the neocortex and hippocampus and reduced number of the cholinergic neurons in basal forebrain and nucleus basalis of Meynert occurs that are closely associated with cognitive deficits in AD (Giacobini, 1997). Pharmacological interventions that enhance acetylcholine levels or block further fall in ACh levels and thus, improve cholinergic neurotransmission are known to produce improvement in learning and memory in AD (Giacobini, 2004). In this context it is interesting to note that acetylcholine has anti-inflammatory actions, and hence, a decrease in the levels of ACh may further aggravate the inflammatory process and progression of AD. This “cholinergic anti-inflammatory pathway” mediated by ACh acts by inhibiting the production of TNF, IL-1, MIF, and HMGB1 and suppresses the activation of NF-êB expression (Borovikova et al., 2000; Pavlov and Tracey, 2004; Wang et al., 2004; Czura et al., 2003).
There is an urgent need for biomarkers to diagnose neurodegenerative disorders early in like AD, when therapy is likely to be most effective, and to monitor responses of patients to new therapies. As research related to this need is currently most advanced for Alzheimer’s disease, Shaw et al., (2007) reviewed the focuses on progress in the development and validation of biomarkers to improve the diagnosis and treatment of AD and related disorders. It is evident form the preceding discussion that presenilin-1 (PS-1), presenilin-2 (PS-2), and amyloid precursor protein (APP) play a significant role in the pathogenesis of Alzheimer’s disease. Missense mutations in APP, PS-1, and PS-2 genes could alter the proteolysis of APP and increase the generation of Aβ42, whose accumulation as diffuse plaques triggers the inflammatory responses due to microglial activation and release of pro-inflammatory cytokines. This is supported by the observation that plasma and cerebrospinal fluid levels of pro-inflammatory cytokines: interleukin-1 (IL-1) and tumor necrosis factor-á (TNF-á) are increased in patients with Alzheimer’s disease (Cacabelos et al., 1991; Fillit et al., 1991; Chao et al., 1994). Systemic injection of IL-1 decreased extracellular acetylcholine in the hippocampus suggesting that increased concentrations of IL-1 in patients with Alzheimer’s disease could be responsible for lowered cerebral acetylcholine levels seen. In addition, IL-1 stimulates the beta-amyloid precursor protein promoter, which is processed out of the larger amyloid precursor protein (APP), which is found in the form of amyloid plaques in the brains of Alzheimer’s diseased patients. Furthermore, receptors of IL-1 are on APP mRNA positive cells and its ability to promote APP gene expression suggests that IL-1 plays an important role in Alzheimer’s disease (Donnelly, 1990; Blume and Vitek, 1989). The involvement of inflammatory process in the pathogenesis of Alzheimer’s disease is further supported by the observation that inhibition or neutralizing the actions of TNF-á could be of benefit to these patients (Tobinick et al., 2006; Rosenberg, 2006). These evidences and the results of the bioinformatics study reported here strongly suggest that PS-1, PS-2 and APP play a dominant role in the pathogenesis of AD by inducing a pro-inflammatory state.
Authors Thankful to partial financial support from IIT up gradation grants of AUC E(A).