Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Review Article - (2015) Volume 0, Issue 0
Biomarkers discovery is a discipline achieving increasing importance since it provides diagnostic/prognostic markers and may permit to investigate and understand the mechanism of development of the pathology, possibly suggesting new biomolecular therapeutic targets. Biomarkers discovery in proteomics is hampered by the use of high-throughput techniques providing a great number of candidates among which the true biomarkers have to be searched for. Moreover, often a small number of samples are available. Two main problems arise when biomarkers have to be searched for in such datasets: 1) the identification of reliable markers, avoiding false positives due to chance correlations; 2) the exhaustive identification of all candidate markers, to obtain a complete snapshot of the effect investigated.
Biomarkers can be identified by two approaches: classical monovariate methods, where each biomarker is considered as independent (Student’s t-test, Mann-Whitney test etc.) or multivariate methods, able to take into consideration the correlation structure of the data (i.e. interactions). These last ones are certainly to be preferred and should achieve the best compromise between the best predictive ability (accomplished through the use of variable selection procedures and exhaustivity. Here, we review the most recent applications of multivariate methods for the identification of biomarkers in proteomics with particular regard to the statistical methods exploited.
Keywords: Biomarker identification; Multivariate statistical methods; Variable selection procedures; PCA; PLS-DA; Random forests; SVM; Ranking-PCA
The field of biomarker discovery has recently developed as one of the most challenging areas of research. Different and heterogeneous disciplines involve the study of biomarkers: clinical and environmental chemistry, ecotoxicology, food research, plant and animal biology. In general, we can speak of biomarkers identification whenever a pool of features differentiating two or more groups of samples has to be identified. For what concerns the particular application to proteomics, the search for biomarkers involves the identification of features responsible for sample differentiation from a quite wide range of instrumental applications: from classical proteomics [1-23], exploiting 2D-PAGE and 2D-DIGE, to mass spectrometry-based approaches based on MALDI-TOF [24-30] and SELDI-TOF profiling [31-42], HPLC-MS [43-57] or shotgun approaches [58,59].
All these heterogeneous applications have a common denominator: all of them are characterized by a great number of variables characterizing each sample, among which biomarkers have to be searched for. Moreover, the great number of variables is often accompanied by a small number of samples available. Two main problems therefore arise when biomarkers have to be identified in such datasets:
- The identification of reliable markers, avoiding false positives, as is the case when chance correlations occur;
- The exhaustive identification of all candidate markers, necessary to obtain a complete snapshot of the effect investigated (a disease, a drug effect, a ripening effect etc).
In all the areas of biomarker search, the final datasets in which biomarkers are search for are characterised by a series of samples divided in two or more classes and described by a series of variables or features. These datasets can be evaluated by the two different strategies:
1) classical statistical methods that identify significant biomarkers by monovariate statistical tests where each biomarker is considered as independent from the others;
2) multivariate methods, able to take into consideration the correlation structure of the data and the synergies and antagonisms (i.e. interactions) existing among the potential biomarkers. This approach is certainly more effective: it generally ensures diagnostic and prognostic performances superior to single markers in terms of sensitivity, specificity and reliability.
The classic approach to the identification of biomarkers involves the evaluation of the variables showing a statistically significant different behavior between two groups of samples (e.g. control vs. pathological, etc.) by classical statistical tests, applied to each biomarker candidate separately, focused to the evaluation of the type I error comparison wise (for each hypothesis independently) or experiment wise (testing all hypotheses together). The second alternative is certainly to be preferred since the type I error probability increases with the number of tests (for k hypotheses=(1-α)k). Usually Student’s t-tests are applied applying a correction that takes into account the number of multiple tests available (Bonferroni’s with subsequent modifications [60], Dunn’s, Sikak’s and Dunnet’s [61-63]). Also non parametric tests can be applied, like the Mann-Whitney test [60] or procedures based on the analysis of variance both in its two-way (ANOVA) or multi-way (MANOVA) versions [60]. MANOVA allows in particular comparing more than two groups of samples. All these strategies have the disadvantage of considering each variable independently from the others so that the correlation structure of the data is not taken into account. This is an important drawback since proteomic datasets are usually quite correlated and groups of molecules showing similar or opposite behavior can be easily encountered, as they often belong to common biochemical pathways where they are transformed into- or react between- themselves.
Multivariate methods instead compare two or more groups of samples considering the relationships existing between the candidate molecules, i.e. the synergic or antagonistic effects of different factors. They are usually coupled to variable selection procedures [16,17] to provide a reduced set of candidate biomarkers with the best predictive ability; standard variable selection tools are in fact aimed to the identification of the most exiguous set of variables with the best predictive ability. It is important to point out that biomarkers are useful not only for diagnostic purposes but also for functional studies to investigate the mechanism of action of a disease or of a particular effect (e.g. ripening, pollution, etc.): from this point of view, highthroughput techniques provide a lot of information that should not be neglected to provide a complete view of the investigated effect. It is therefore the authors’ opinion that exhaustivity should be addressed to as well [64,65].
Some reviews have recently been published on biomarker discovery in several fields of proteomics: food and beverages [66], toxicology [67], molecular plant physiology [68], clinical proteomics with the particular application to colorectal cancer [69], Alzheimer’s disease [70], traumatic brain injury [71], type-1 diabetes [72], urine molecular profiling [73,74]. Other authors instead reviewed the application of instrumental or statistical tools for biomarkers identification in proteomics: LC-MS [75], MALDI [76], machine learning algorithms [77] and statistical data processing in clinical proteomics [78].
Here, we review the most recent applications of multivariate statistical methods for the identification of biomarkers in proteomics with a particular attention to the statistical methods exploited. Literature applications are grouped by the exploited multivariate methods; a brief description of the theoretical aspects of each method is presented at the beginning of each paragraph.
Multivariate methods compare two or more groups of samples considering the relationships existing between the variables (candidate molecules): this corresponds to considering their synergic or antagonistic effects, i.e. their interactions. In the field of biomarkers identification, these methods usually belong to one of these categories:
1) unsupervised pattern recognition methods, also called clustering methods, based on no a priori information: the evaluation of the existence of groups of samples is suggested by the statistical method itself;
2) supervised classification methods, based on a priori information about the membership of each sample to a specific class: aimed to the identification of the variables responsible for the separation of the samples in the different classes.
All methods are applied to datasets where each sample is described by a series of variables: spot volumes in datasets from 2D-PAGE and 2D-DIGE or peak intensities in mass spectrometry-based approaches. It is important to point out that multivariate methods can be applied to all the quantitative proteomics analysis, a fundamental requirement for biomarker discovery analysis. Here, only the methods most recently applied in literature in the proteomic field will be discussed. Table 1 reports all the main statistical methods used in proteomics.
| Type of method | Method family | Method adopted |
|---|---|---|
| Unsupervised pattern recognition methods | Methods based on data projection to other dimensions | Principal Component Analysis (PCA) |
| Nonnegative PCA | ||
| Methods based on correlation analysis | Canonical Correlation Analysis (CCA) | |
| Sparse Canonical Correlation Analysis (SCCA) | ||
| Cluster analysis methods | Hierarchical Clustering | |
| Supervised classification methods | Methods based on PCA | Soft-Independent Model of Class- Analogy (SIMCA) |
| Principal Component Analysis – Discriminant Analysis (PCA-DA) | ||
| Ranking-PCA | ||
| Bayesian methods | Linear Discriminant Analysis (LDA) | |
| Diagonal LDA (DLDA) | ||
| Methods based on projection to latent variables | Partial Least Squares Discriminant Analysis (PLS-DA) | |
| Orthogonal Partial Least Squares (OPLS) | ||
| Classification trees | Classification and Regression Tree (CART) | |
| Random forests (RF) | ||
| Machine learning | Support Vector Machines (SVM) |
Table 1: Unsupervised and supervised methods for biomarker identification.
Classification performance
Classification can be evaluated by representing the classification matrix C, where true classes are reported on rows and assigned classes on columns; correct classifications are therefore reported along the matrix diagonal (elements cgg), while wrong assignments are reported in extra-diagonal elements (cij, i≠j). From the classification matrix it is possible to calculate some parameters accounting for the performance of classification:
Non-error rate (NER%): the percentage of overall correct assignments:
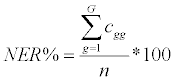
Where, cgg are the diagonal elements of the classification matrix; n is the overall number of objects; G is the overall number of classes.
Selectivity Sg: the percentage of non-overlap between the classes:
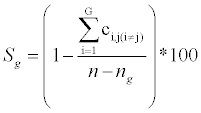
Where, ci,j are the extra-diagonal elements (the sum runs on the rows of the classification matrix); n is the number of overall objects; ng is the number of objects in class g; G is the overall number of classes.
Specificity Spg: the NER% of each class:
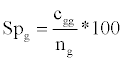
Where, cgg is the number of correct classification of class g; ng is the umber of objects of class g.
These parameters can be calculated both in calibration and crossvalidation for obtaining an evaluation of the predictive ability of the classification model.
Unsupervised pattern recognition methods
Several applications of pattern recognition methods are present in literature: in the most of papers Principal Component Analysis (PCA) and/or hierarchical clustering are applied both to classical proteomics (2D-PAGE or 2D-DIGE maps) and to mass spectrometric data from direct sample analysis by MALDI-TOF, SELDI-TOF or HPLC-MS. Both these approaches can be applied to all quantitative analytical methods exploited in proteomics and show no strict constraints on the number of variables and/or samples that have to be present in the dataset: in the case of PCA, when a smaller number of samples than of variables is present, the maximum number of PCs that can be calculated equals the number of samples present.
Principal Component Analysis (PCA): PCA is by far the most widespread pattern recognition tool in proteomics: in this section a selection of the applications regarding exclusively PCA or hierarchical clustering will be presented, a more exhaustive list being reported in table 2.
| Field | Sample | Study | Analytical method | Statistical method | Ref |
|---|---|---|---|---|---|
| Ecotoxicology and environmental chemistry | Mussels | 2D-PAGE | PCA | [81] | |
| Plasma samples of Atlantic cods | Exposure to alkylphenol | SELDI-TOF MS | PCA | [31] | |
| Plasma samples of Atlantic cods | Exposure to estrogens | SELDI-TOF MS | PCA | [32] | |
| Sydney Rock oysters | Exposure to metal contamination | 2D-PAGE | PCA | [2] | |
| Digestive gland of the sentinel "blue mussel" | Assessment of marine pollution | Cell fractionation followed by ion-exchange chromatography and 2-DE | Hierarchical clustering | [14] | |
| Hake liver and brain extracts | Investigation of population variability | 2D/DIGE and MS | Hierarchical clustering | [15] | |
| Clinical biomarker discovery | Serum samples | Differential protein biomarker expression and their time-course in coronary syndromes | Microarray | PCA | [83] |
| Serum samples | Influence of clotting time on the protein composition | Label-free and stable-isotope labeling MS | PCA | [45] | |
| Plasma | Identification of biomarkers in patients with acute myocardial infarction | 2-D-DiGE | PCA | [4] | |
| Inflamed and non-inflamed colon biopsies | Identification of markers for ulcerative colitis | 2D-gel electrophoresis and MALDI-TOF MS | PCA | [5] | |
| Serum | Protein and peptide profiling in depression | MALDI-MS | PCA | [25] | |
| Serum and pleural effusion | Identification of markers for non-small cell lung cancer | 2D-DIGE | PCA | [3] | |
| Cell lines | Identification of markers for non-small cell lung cancer | 2D-DIGE | PCA | [9] | |
| Plasma | Effect of a high intake of industrial or ruminant trans fatty acids on healthy men | 2DE | PCA | [8] | |
| Brain samples of rats | Peptide profiles of exposition to amphetamine | MALDI-TOF MS | PCA | [26] | |
| Biopsies | Identification of markers for thyroid proliferative diseases | SELDI-TOF-MS | PCA | [34] | |
| Plasma | Identification of markers for disease progression for idiopathic pneumonia syndrome following allogeneic stem cell transplantation | LC-MS | PCA | [84] | |
| Pancreatic ductal carcinoma cells | Effect of Trichostatin A | 2D-PAGE | PCA | [20] | |
| Body fluids | Development of the analytical method | SOMAmers | PCA | [85] | |
| Plasma | Identification of markers in a mouse intestinal tumor model | LC-MS/MS | Hierarchical clustering | [50] | |
| Serum | Peptidomics in Crohn's disease | Label-free nano-HPLC-MS | Hierarchical clustering | [43] | |
| Liver | Biomarkers for progressive alcoholic steatosis | 2-DE | Hierarchical clustering | [10] | |
| Tissues | Different cancers | MALDI imaging | Hierarchical clustering | [88] | |
| Tissues | Characterization of the proteomic changes in Barrett's adenocarcinoma and its premalignant stages | MALDI imaging | Hierarchical clustering | [89] | |
| Assessing measurement reproducibility in biomarker research | Reference human serum standard | Evaluate the measurement reproducibility | SELDI-TOF | PCA | [33] |
| Serum of cervical cancer patients | Development of an improved sample preparation method to perform future comparative analyses of samples from a bank of patients | LC-MS | PCA | [44] |
Table 2: Applications of pattern recognition methods in proteomics.
PCA [79,80] represents the objects, described by the original variables, in a new reference system characterised by new variables called Principal Components (PCs). PCs are calculated hierarchically: the first PC accounts for the maximum variance contained in the original dataset, while subsequent PCs account for the maximum residual variance; in this way systematic variations are explained in the first PCs while experimental noise and random variations are contained in the last ones. The PCs are linear combinations of the original variables. They are also orthogonal to each other, thus accounting for independent sources of information. A graphical example is presented in Figure 1a, where PCs are calculated in a simple case where only two original variables X1 and X2 are present. PCs are often used for dimensionality reduction due to their hierarchical nature: the original variables can be substituted by a smaller number of significant PCs, containing only relevant information. PCA provides two main tools for data analysis (Figure 1b):

Figure 1: PCA calculation: graphical representation of PCs (a) and calculation of a PCA model (b).
- the scores, representing the co-ordinates of the samples in the space given by PCs;
- the loadings, representing the coefficients of the linear combination describing each PC, i.e. the weights of the original variables on each PC.
The samples are usually graphically represented in the space given by the PCs (through their scores) to allow the identification of groups of samples showing similar (samples close one to the other in the graph) or different (samples far from each other) behaviours. By looking at the corresponding loading plot (where the weights of each original candidate molecule on each PC are represented), it is possible to identify the variables that are responsible for the similar or different behaviours detected for the samples in the score plot.
In the most of applications PCA is exploited for the visualization of the results in terms of separation achieved for the different classes in the space given by the relevant PCs. However, PCA provides important information worth of being exploited for data interpretation as well: the loadings provide information on the candidate biomarkers and their up- or down- regulation in the investigated case study; some applications are also present that exploit this information for the identification of the most relevant biomarkers.
Some applications regard the identification of biomarkers in ecotoxicological studies: for the biomonitoring of mussels after the Prestige’s oil spill by 2D-PAGE [81]; to identify biomarkers of exposure to alkylphenol [31] and estrogens [32] in plasma samples of Atlantic cods by SELDI-TOF MS; to study the proteomic pattern of Sydney Rock oysters exposed to metal contamination by 2D-PAGE [2].
However, the majority of applications is in the field of clinical biomarker discovery to investigate (Table 2): a) coronary syndromes [82]; b) the influence of clotting time on the protein composition of serum samples [45]; c) acute myocardial infarction [4] d) inflamed and non-inflamed colon biopsies [5] in ulcerative colitis depression [25]; e) non-small cell lung cancer [3,9]; f) whether a high intake of industrial or ruminant trans fatty acids affects the plasma proteome [8]; g) amphetamine in rats [26]; h) thyroid proliferative diseases [34]; i) idiopathic pneumonia syndrome following allogeneic stem cell transplantation [83]; j) the effect of Trichostatin A on pancreatic ductal carcinoma cells [20]; the development of a special class of aptamers [84], called SOMAmers (slow off-rate modified aptamers), which bind specifically to proteins in body fluids.
Other applications regard the use of PCA as a tool for assessing measurement reproducibility in biomarker research: Liggett et al. [33] applied PCA to SELDI-TOF profiles of repeated measurements of a reference human serum standard, while Govorukhina et al. [44] developed an improved sample preparation method to perform future comparative analyses of samples from a serum bank of cervical cancer patients.
Cluster analysis and hierarchical clustering: Cluster analysis techniques [79,80,85] allow the identification of groups of samples or of descriptors in a dataset. The most used clustering methods belong to the class of the agglomerative hierarchical methods [79,80], where the samples are grouped (linked together) on the basis of a measure of their similarity. The most similar samples or groups of samples are linked first. The final result is a graph, called dendrogram, where the samples are represented on the X axis and are connected at decreasing levels of similarity along the Y axis. The groups can be identified by applying a horizontal cut of the dendrogram, i.e. at a particular level of dissimilarity, and identifying the number of vertical lines crossed by the horizontal cut. In Figure 2 it is reported the example of a dendrogram obtained from a 2D-PAGE spot volume dataset: cutting at level 2000 identifies two clusters, corresponding to 2 different human pancreatic tumour cell lines, T3M4 and PACA44, while cutting at level 4500 identifies 4 clusters (the two cell lines treated and untreated with Trichostatin-A). The results of hierarchical clustering strongly depend on the specific measure of similarity and on the linking method adopted. Clustering techniques can be applied either to the original variables or to the scores of the relevant PCs thus achieving clustering exploiting only useful sources of variation, being the experimental error eliminated in the last PCs [85]. In two-way hierarchical clustering a graphical representation of clustering of both variables and samples is provided, to visually identify cluster of samples and in the meantime provide information on the behaviour of the variables in the different clusters. This particular application is quite widespread in both proteomics and genomics.

Figure 2: Dendrogram calculated by the Ward’s linkage method and exploiting euclidean distances. Samples, reported on the x-axis, belong to two pancreatic cancer cell lines (T3M4 and PACA44) treated or not with Throchostatin-A (TSA).
Hierarchical clustering has been applied in combination to PCA in a series of papers focused to the identification of biomarkers in clinical and environmental applications: to identify groups of samples with a similar behavior and/or groups of variables with a similar expression. In some cases two-way hierarchical clustering was applied [13,86], to identify groups of samples and at the same time provide information about the behavior of the variables in the identified groups. Also in this case, hierarchical clustering can be applied to data from classical proteomics by 2D-PAGE or 2D-DIGE, or to mass spectrometric data from MALDI-TOF, SELDI-TOF or HPLC-MS.
The most of applications (Table 2 for more details) are in the field of clinical proteomics to investigate: a) ovarian [49], prostate [11] and colorectal cancer [12]; b) systemic and invasive candidiasis [13,86]; c) the plasma proteome in a mouse intestinal tumor model [50]; d) serum peptidomics in Crohn’s disease [43]; e) liver proteomics in progressive alcoholic steatosis [10].
Two applications regard MALDI imaging: in the first study Deininger et al. [87] applied MALDI imaging to compare spectra from controls and patients affected by different cancers. The reconstruction of images based on PC scores allowed an unsupervised feature extraction of the dataset. Generally, these images were in good agreement with the histology of the samples. The hierarchical clustering allowed the access to the multidimensional information in the dataset and the selection of spectra classes representative for different tissue features. PCA showed that the tumor and control mucosa were separated in the first three PCs. In the second study Elsner et al. [88] applied hierarchical clustering to MALDI imaging MS results, to characterize proteomic changes found in Barrett’s adenocarcinoma and its premalignant stages and find proteins that might be used as markers for monitoring cancer development as well as for predicting regional lymph node metastasis and disease outcome.
Two papers regard applications to environmental analysis for the assessment of marine pollution on blue mussels [14] and for evaluating the protein expression in liver and brain extracts of hakes [15].
Supervised classification methods
Supervised classification tools are used to separate the objects in the classes present which are known a priori (e.g. control versus pathological) and provide the variables most responsible for their belonging to different classes (candidate biomarkers). Their application in the proteomics field is focused both to the development of diagnostic tools and to the identification of the differences existing between the classes, to shed light on the mechanism of action of the effect under investigation (e.g. a disease or a new drug in the biomedical field, ripening in food research, a cultivar in plant biology, etc.). Here, only the methods already applied to the identification of biomarkers in the proteomics field will be presented (Table 1).
All the presented methods can be applied to all quantitative analytical methods exploited in proteomics. Methods based on PCA or on projection to latent structures (as PLS-DA and OPLS) show no strict constraints on the number of variables and/or samples that have to be present in the dataset: they provide a substantial dimensionality reduction and, when a smaller number of samples than of original variables is present, the maximum number of latent structures that can be considered equals the number of samples. Other methods, as Linear Discriminant Analysis (LDA) can be applied when the number of variables overcomes the samples available: when this constraint is not observed, LDA can be applied to PCs or coupled to variable selection procedures, giving a final model containing a maximum number of original variables equaling the number of samples.
Several papers have recently appeared on the development and application of classification tools to proteomic datasets (2DE, 2D-DIGE, MALDI-TOF, SELDI-TOF, HPLS-MS data). These tools represent certainly a better approach than the use of pattern recognition methods since they provide mathematical models able to clearly identify sets of candidate biomarkers and provide information on classification performances. In the field of proteomics, a great panorama of different techniques have been recently developed and applied: some of the most significant papers in this field will be presented here separated according to the classification tool exploited in the study, a more exhaustive list of applications being presented in table 3.
| Field | Sample | Study | Analytical method | Statistical method | Performance | Ref |
|---|---|---|---|---|---|---|
| Clinical proteomics | Sera | Gaucher patients and healthy controls | SELDI-TOF | PCA-DA with variable selection by rank products | Sg 89%; Spg 90% | [91] |
| Sera | Breast cancer patients and healthy controls | MALDI-MS | PCA-DA with variable selection by rank products | Sg 82%; Spg 86% | [92] | |
| Ecotoxicology | Mussels | Exposition to oil pollution | SELDI-TOF | CART | - | [53] |
| Clinical proteomics | Proximal fluid samples | Identify biosignatures of 3 breast cancer types: HER2 positive, hormone receptor positive and HER2 negative, triple negative (HER2-, ER-, PR-). | Protein fractionation before LC-MS/MS | CART | - | [54] |
| Sera | Characterization of the response to Infliximab in Crohn's disease: 20 patients with or without clinical response to Infliximab | SELDI-TOF-MS | CART | Sg, Spg and accuracy in cross-validation: 78.6%, 80.0%and 79.3% |
[35] | |
| Sera | Hepatocellular carcinoma: 81 patients with hepatitis B-related carcinoma and 80 controls | SELDI-TOF | CART | Sgand Spg89.6%. Model with two biomarkers and AFP: Sg91.7%; Spg92.7%. | [39] | |
| Urine | Predictive diagnosis of chronic allograft dysfunction: 29 samples withdrawn 3 months post-transplant | SELDI-TOF | CART | Sg 93%; Spg65%. | [36] | |
| Clinical proteomics | Platelets, peripheral blood mononuclear cells, plasma, urine and saliva | Investigation of how fasting for 36h, as compared to 12h, affects the proteome of healthy volunteers | 2DE, MS and multiplex immunoassay | Random forests | - | [6] |
| Sera | Biomarker for Malignant Pleural Mesothelioma: 117 pathological cases and 142 asbestos-exposed controls | SOMAmer proteomic technology | Random forests | Sg: 97% (training) – 90% (blinded verification); Spg 92% (training), 95% (blinded verification). Second validation set: Sg/Spg 90%/89%; combined accuracy 92%. | [106] | |
| Plasma | Biomarkers for multiple systemic autoimmune diseases in disease-discordant monozygotic twins: 4 pairs of systemic lupus erythematosus, 4 pairs of juvenile idiopathic arthritis, 2 pairs of juvenile dermatomyositis | RP-LC-MS | Random forests | - | [57] | |
| Sera | Identification of lymphnode metastases in NSCLC with circulating autoantibody biomarkers | 2-D immunoblots of HCC827 lysates for tumor-associated autoantigens | Random forests | Sg94%, Spg97%, NER% 96% | [107] | |
| Blood | NSCLC | Immunoproteomic method | Random forests | NER%: 97% | [108] | |
| Sera | Biomarkers for prostate cancer | 2D-DIGE | Random forests | - | [19] | |
| Clinical proteomics | Cytosolic protein extracts from frozen thyroid samples | Biomarkers for follicular and papillary thyroid tumors: 10 follicular adenomas, 9 follicular carcinomas, 10 papillary carcinomas, 10 controls | 2DE | PLS-DA | - | [18] |
| Urine | Peptidomics | LC/MS | PCA and PLS-DA | - | [98] | |
| Sera | Biomarkers of ovarian cancer: 265 sera from women admitted with symptoms of a pelvic mass | MALDI-MS | PCA and PLS-DA | Best models: 79% Spg, 56% Sg, 68% accuracy | [95] | |
| Cell line extracts | Biomarkers for colon cancer (HCT116 cell line) treated and not treated with a new histone deacetylase inhibitor | 2D-PAGE | PLS-DA | NER%=100% | [17] | |
| Sera | Biomarkers of resistance to neoadjuvant chemotherapy in advanced breast cancers: profiling of N-glycosylated proteins in 15 advanced breast cancer patients | Label-free LC-MS/MS | PLS-DA | - | [56] | |
| Cerebrospinal fluid | Markers of multiple sclerosis (MS) and other neurological diseases (OND) vs. controls (NHC) | Mas spectral profiling | PLS-DA | NER%: MS vs OND: MS 89.5%, OND: 92.3%. MS vs NHC: 100%. OND vs NHC: OND 97.2%, NHC 98.4% |
[96] | |
| Plasma | Biomarkers of Alzheimer's disease progression: 119 samples of patients with mild cognitive impairment (MCI) with different outcomes | Untargeted, label-free shotgun proteomics | OPLS-DA | Best model: accuracy 79%. Some sex-specific biomarkers were identified. | [58] | |
| Clinical proteomics | Sera | Biomarkers of cancer (lymphoma and ovarian): determination of N-glycans of human serum alpha-1-acid glycoprotein | MALDI-TOF MS | LDA | NER% 88%. Cross-validation: cancerous vs. controls Sg 96%, Spg 93%; lymphoma vs. controls + ovarian tumor 72% Sg 84% Spg | [28] |
| Sera | Development of a novel index FI-PRO in the prediction of fibrosis in chronic hepatitis C: 62 patients for training and 73 for validation. Prediction of minor fibrosis (F0-F1), moderate fibrosis (F2-F3) and cirrhosis (F4). | - | LDA | Best model based on four markers. Novel index A2M/hemopexin: diagnostic performance rate 0.80-0.92 for F2-F4 and F3-F4 in validation | [104] | |
| Plant biology | Pinot Noir skins | Biomarkers of ripening: 3 moments of ripening | 2DE | PCA and LDA | NER%=100% in calibration; 77.78% in cross-validation | [16] |
| Animal biology | Sera | Biomarkers of ovine paratuberculosis (Johne's disease): sheep with paratuberculosis, vaccinated-exposed sheep and unexposed animals | SELDI TOF–MS | CART and LDA | Accuracy: sheep vs unexposed or exposed 75-100% | [38] |
| Clinical proteomics | Simulated data and a proteomic dataset | Development of Ranking-PCA | 2-DE | Ranking-PCA | NER%=100% | [65] |
| Differet samples | Three different proteomic datasets: 1) 8 2DE maps from adrenal nude mouse glands (4 controls and 4 affected by neuroblastoma); 2) 11 samples from nuclea of human colon cancer HCT116 cell line (6 controls and 5 treated by an HDAC inhibitor); 3) 10 samples from total lysates of human colon cancer HCT116 cell line (5 controls and 5 treated by an HDAC inhibitor) | 2-DE | Ranking-PCA | NER%=100% | [64] | |
| Food analysis | Meat extracts | Biomarkers of tenderization of bovine Longissimus dorsi: 4 Charolaise heifers and 4 Charolaise bull’s muscles sampled at slaughter after early (12 days) and long ageing (26 days) | Cartesian and polar 2-DE | Ranking-PCA | NER%: 100% | [90] |
| Clinical proteomics | Cell line extracts | Biomarkers for neuroblastoma | 2-DE | SIMCA | NER%: 100% | [21] |
| Cell line extracts | Biomarkers of mantle cell lymphoma | 2DE | SIMCA | NER%: 100% | [22] | |
| Cell line extracts | Development of an approach for identifying relevant proteins from SIMCA DPs | 2DE | SIMCA | NER%: 100% | [23] | |
| Clinical proteomics | Sera | Development of a sequence-specific exopeptidase activity test. Application to metastatic thyroid cancer patients (48) and controls (48) | MALDI-TOF MS | SVM | 94% Sg and 90% Spg | [29] |
| Plasma | Biomarkers of air contaminant exposure: Fischer rats exposed for 4h to clean air or Ottawa urban particles | HPLC with autofluorescence detectio | SVM and GA | - | [113] | |
| Sera | Diagnosis of gastric adenocarcinoma. Test/training set: 120 gastric adenocarcinoma and 120 controls. Validation: 95 gastric adenocarcinoma and 51 controls. | 29-plex array platform | Random forests and SVM | Training/test set: accuracy >88%. Validation set: >85%. | [114] | |
| Cerebrospinal fluid | Biomarkers of multiple sclerosis-related disorders: 107 patients with MS-related disorders (including relapsing remitting MS [RRMS], primary progressive MS [PPMS], anti-aquaporin4 antibody seropositive-neuromyelitis optica spectrum disorder [SP-NMOSD], and seronegative-NMOSD [SN-NMOSD]), amyotrophic lateral sclerosis (ALS), other inflammatory neurological diseases (controls). Independent sample set of 84 patients with MS-related disorders or with other neurological diseases. | MALDI-TOF MS | PCA and SVM | SP-NMOSD and SN-NMOSD distinguishable from RRMS with high cross-validation accuracy by SVM | [30] | |
| Sera | Biomarkers of NSCLC: 8 NSCLC samples and 8 controls | Label-free quantitative 1D-LC/MS/MS | Normalized, randomly paired t test and integrated bioinformatics, including hierarchical clustering analysis, PCA and SVM | - | [59] | |
| Plasma | Biomarkers of tuberculosis and malaria | SELDI-TOF and MS | SCCA and SVM | Improvementsin diagnostic prediction, up to 11% in tuberculosis and up to 5% in malaria | [112] | |
| Urine | Biomarkers associated with early renal injury: 50 healthy controls and intensive care unit patients 12 - 24h after coronary artery bypass graft surgery | SELDI-TOF MS | SVM coupled to PCA | - | [37] | |
| Plasma | - | 2-D-LC-MS | Regression analysis, unsupervised hierarchical clustering, PCA, genetic algorithm and SVM | 88% Sg and 94% Spg | [51] | |
| Clinical proteomics | Sera | Identification of discriminatory variables in MS by clustering of variables (CLoVA). Two experimental data sets: ovarian and prostate cancers. | MALDI-TOF and SELDI-TOF | Self-organization maps for clustering of variables; classification methods: PLS-DA and ECVA | Higher Sg and Spg than conventional PLS-DA and ECVA | [115] |
| Plasma | Identification of a liver cirrhosis signature for predicting hepatocellular carcinoma risk in Hepatitis B carriers | 174-antibody microarray system | PCA, DLDA and 3-NN | Accuracy, Sg and Spg: 100%, 100% and 90.9% respectively |
[119] | |
| Plasma | Biomarkers for depression and schizophrenia: 245 depressed patients, 229 schizophrenic patients and 254 controls | Multi analyte profiling evaluating 79 proteins | PCA, PLS-DA and random forests | - | [99] | |
| Urine | Biomarkers of pediatric nephrotic syndrome (NS): steroid-sensitive NS (SSNS), steroid-resistant NS (SRNS), and orthostatic proteinuria (OP). 19 subjects with SSNS/SDNS in remission, 14 with SSNS/SDNS in relapse, 5 with SRNS in relapse, and 6 with OP. | SELDI-TOF MS | Genetic algorithm and PCA | - | [40] | |
| Sera | Evaluation of intact alpha-1-acid glycoprotein isoforms as potential biomarkers in bladder cancer: 16 samples (8 healthy, 8 bladder cancer) | CZE-UV and CZE-ESI-MS | ANOVA, PCA, LDA and PLS-DA. | Best results obtained by LDA: NER%=93.75% | [127] | |
| Tear fluid | Biomarkers of breast cancer: 50 women with breast cancer and 50 age-matched controls | SELDI-TOF MS | multivariate discriminant analysis and ANN | NER%: 71.19% for cancers, 70.69% for controls (overall NER=70.94%) | [42] | |
| Urine | Two studies: 1) addition of seven peptides at nanomolar concentrations to blank urine samples of different origin; 2) a study of urine from kidney patients with and without proteinuria. | LC-MS | PCA and NSC | - | [46] | |
| Plant biology | Leaves of Arabidopsis thaliana |
Analysis oftime-related regulatory effects of plant metabolism at a systems level: wild type plants and starchless mutant plants deficient in phosphoglucomutase activity | GC-TOF-MS- metabolite profiling and LC-MS- protein profiling | PCA and ICA | - | [47] |
| Clinical proteomics | Sera and plasma | Biomarkers of inflammatory auto-immune disease: 30 patients | MALDI-TOF | ICA | - | [24] |
| Maternal plasma and cord plasma | Biomarkers of spontaneous preterm birth: 191 African, American and Caucasian women | - | MARS | - | [121] | |
| - | Improvement of mass spectra classification | MALDI-TOF or SELDI-TOF | MCR | - | [27] | |
| Plasma and bone-marrow cell extracts | Biomarkers of acute myeloid or acute lymphoblastic leukemia: patients with Kawasaki disease and bone-marrow cell extracts from patients with acute myeloid or acute lymphoblastic leukemia | SELDI-TOF-MS | Preprocessing algorithm that clusters highly correlated features, using the Bayes information criterion to select an optimal number of clusters | - | [116] | |
| Proteomic datasets of ovarian and prostate cancer | Development of a new approach to biomarker selection based on the application of several competing feature ranking procedures to compute a consensus list of features | SELDI-TOF | random forest, SVM, CART, LDA | - | [117] | |
| Sera | Development of Nonnegative PCA. Four serum proteomic datasets: ovarian, ovarian-qaqc (quality assurance/quality control), liver and colorectal |
MS profiling | nonnegative PCA and SVM | [81] | ||
| Sera | Biomarkers of Type 1 diabetes (T1D) | SELDI-TOF | Normal kernel discriminant analysis | Training set: 88.9% Spg, 90.0% Sg. Test set: 82.8% Spg, 76.2% Sg | [41] |
Table 3: Applications of supervised methods in proteomics.
Principal Component Analysis–Discriminant Analysis (PCADA): PCA-DA was first developed by Hoogerbrugge et al. [89]. In this method PCA is exploited as a dimensionality reduction tool. The space defined by the relevant PCs is used to identify linear discriminant functions (LDFs) able to separate the samples in the classes present. The first LDF, D1, is defined as:
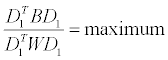
where:
B=between group covariance matrix;
W=within group covariance matrix.
The second LDF, D2, is defined in the same way but under the condition that D1 and D2 are independent. In two class problems, only one LDF is provided, able to discriminate the samples in the two classes present.
An interesting paper appeared by Smit et al. [90] presenting a strategy for the statistical validation of discrimination models in proteomic studies by PCA-DA applied to SELDI-TOF analyses of serum samples. Different tools as permutation tests, single and double crossvalidation are combined to provide a statistically sound procedure for biomarker discovery. The cross-validation steps were combined with a variable selection method called rank products. The strategy was applied coupled to PCA-DA but any other classifier could be used. A dataset containing serum samples from Gaucher patients and healthy controls was used as test case. The variable selection procedure can be briefly described as follows: the discriminant vector found with PCADA represents the differences between the control and the diseased groups. Since the largest peaks in this vector are most important for the discrimination, the m/z values can be selected on the basis of their absolute value in the discriminant vector. In a 10-fold cross-validation, 10 different discriminant vectors are found in which the importance of the m/z values is different. For each of the discriminant vectors, the m/z values are ranked according to their absolute value; the m/z value with the largest absolute value gets rank 1, the next largest gets rank 2, etc. The 10 ranks of each m/z value are multiplied to obtain the rank product, and the m/z values with the lowest rank product are the ones with the largest discriminative power. Single cross-validation in combination with rank products can be used for variable selection, while the prediction error associated with the selected variables is determined with double cross-validation. The model presents Sg=89% and the Spg=90%. The validation of the discrimination models with a combination of permutation tests and double cross-validation helps to avoid erroneous results which may result from undersampling, as it is often the case in proteomics.
The same authors [91] applied the procedure to a dataset containing serum samples from breast cancer patients and healthy controls obtaining Sg=82% and Spg=86%. The final classification exploited a majority voting scheme from the ensemble classifier.
Ranking-PCA is a ranking method proposed by Marengo, Robotti et al. [64,65,92] based on the description of the original data by means of PCs. The development of this method has its roots in the necessity of finding the optimal compromise between best predictive ability of the final model and the exhaustivity of the biomarkers search. PCA is used to describe the data coupled to a ranking procedure of the candidate biomarkers in forward search. When PCA is applied to a dataset where the samples belong to two classes and their belonging to class 1 or 2 is the leading information, should provide only the first PC as relevant for the samples discrimination. When the variable ranking procedure is applied in forward search, one variable is added at each cycle. The first variable selected is the one providing the best separation between the classes on the first PC (Figure 3a). The addition of another discriminating variable further improves the distance between the two classes on PC1 (Figure 3b). If successively a non-discriminating variable is added, instead, the two classes will not be further separated on PC1: the third variable will show a small weight on the first PC and will be mostly explained by other PCs. However, these subsequent PCs will not be considered relevant since they are not related to class separation. Sometimes, more than one PC could be necessary for class separation (Figure 3c): in this case different independent sources of information related to the class structure are present. A third variable acting in this way could be explained mostly by another PC (Figure 3d) or could show large weight on both PC1 and the other PC responsible for class separation: in both cases, the second PC accounting for class separation will be included in the model. The algorithm is structured in two steps:

Figure 3: Mechanisms of construction of a Ranking-PCA model. Separation of the classes along PC1 after the addition of the first discriminating variable (a). Further addition of another discriminating variable improves classes separation along PC1 (b). Addition of a third variable accounting for another source of variation: scores (c); loadings (d).
1) Selection of the first variable. The first variable is the one providing the largest distance between the two class centroids while preserving class compactness;
2) Selection of the subsequent variables. In the subsequent steps the variable chosen at each step is the one providing the maximum increase of distance between the two centroids in the space given by the relevant PCs, while preserving class compactness.
At each cycle, the possibility of including more than one PC is considered through the exhaustive evaluation of all possible classification models containing from 1 to a user-selected maximum number of PCs. The choice of the most discriminating variable is performed by cross-validation: the final model therefore represents the best model according to its predictive ability. The proposed method allows the ranking of the variables according to their discrimination ability, assuring the exhaustiveness of the results.
The first applications of this method regard the identification of biomarkers from proteomic spot volume datasets. In a first study [65] the authors investigated an artificial dataset and a real casestudy to demonstrate its principle: Ranking-PCA exhaustively identified the potential biomarkers and provided reliable and robust results. In another paper [64] the same method was applied to three different proteomic datasets to prove its effectiveness: 1) 8 2DE maps from adrenal nude mouse glands (4 controls and 4 affected by neuroblastoma) described by 532 spots; 2) 11 samples from nuclea of human colon cancer HCT116 cell line (6 controls and 5 treated by an HDAC inhibitor) described by 779 spots; 3) 10 samples from total lysates of human colon cancer HCT116 cell line (5 controls and 5 treated by an HDAC inhibitor) described by 525 spots. Ranking PCA provided the perfect classification of all samples and provided a more exhaustive identification of biomarkers if compared to previously published results based on the use of other classification tools.
Another application [92] by the same authors regards the study of the proteomic changes involved in tenderization of bovine Longissimus dorsi: 4 Charolaise heifers and 4 Charolaise bull’s muscles were sampled at slaughter after early and long ageing (2-4°C for 12 and 26 days respectively). Protein composition of fresh muscle and of aged meat was analyzed by cartesian and polar 2-D electrophoresis. Ranking-PCA was applied to detect proteomic modulation: meat maturation caused changes of the abundance of proteins involved in metabolic, structural, and stress related processes.
Soft-Independent Model of Class-Analogy (SIMCA)
SIMCA [21-23,93] is based on the independent modelling of each class by means of PCA: each class is described by its relevant PCs. The samples belonging to each class are contained in the so-called SIMCA boxes, defined by the relevant PCs of each class (an example of different SIMCA boxes is presented in Figure 4). Exploiting PCA, the classification of each sample with SIMCA is not affected by experimental uncertainty and random variations since each class is modelled only by its relevant PCs. This method is also useful when more variables than objects are available since it performs a substantial dimensionality reduction.

Figure 4: Example of SIMCA classification: the three classes are described by SIMCA boxes built with one (class A), two (class B) and three PCs (class C); Object * is nearer to class A but it is classified in none of the classes present since it falls outside the three classes boundaries.
The classification rule of object i is based on a Fisher’s F-test; the residual standard deviation of each object i (i.e. its distance from the model of class g) is compared to the residual standard deviation of class g (i.e. the typical distance of class g): if their ratio is smaller than the critical F value based on the degrees of freedom and on the significance level, object i is classified in class g. With SIMCA also outliers can be identified, i.e. samples classified in none of the classes: this happens when an object lies outside all the existing SIMCA boxes (an example is given in Figure 4).
SIMCA provides an important statistics useful for the identification of the most discriminating variables (i.e. candidate biomarkers): the Discrimination Power (DP) which is a measure of the ability of each variable to discriminate between two classes (c and g) at a time. The greater the discrimination power, the more a variable influence the classification of an object to class c or g.
The discrimination power is positive defined, but it is not limited.
SIMCA was applied by Marengo et al. in two studies. The first one [21] is focused on the identification of biomarkers for neuroblastoma the most common extracranial solid tumor of infancy and childhood, by 2D-PAGE. The second study is devoted to identify biomarkers of mantle cell lymphoma [22] by evaluating the different expression of two different human lymphoma cell lines by 2D-PAGE. When SIMCA is applied as classification method, this can be done by the analysis of the discrimination power of each candidate biomarker: usually, variables characterized by a DP larger than a selected threshold are considered significant. The same authors developed an approach [23] for identifying relevant proteins from SIMCA discriminating powers not based on a threshold level established by the operator. The method is based on a procedure consisting in two steps: 1) through a nonlinear Box-Cox transformation, the population of the calculated DP values is turned into a well-known statistical distribution (e.g., Gaussian or gamma) and 2) the relevant spots are identified, by the use of probability plots, as those characterized by a transformed DP value that does not match the reference statistical distribution. The idea underlying the procedure is that the variables characterized by a relevant DP value do not belong to the population of the spots showing homogeneous values of DPs. The method successfully allowed the identification of the relevant spots from 2D maps in several cases study.
PLS [79,80,94] is a multivariate regression method establishing a relationship between one or more dependent variables (Y) and a group of descriptors (X). X and Y variables are modeled simultaneously, to find the latent variables (LVs) in X that will predict the LVs in Y (Figure 5a). These LVs are calculated hierarchically, as for PCA. PLS was originally set up to model continuous responses but it can be applied even for classification purposes by establishing an appropriate Y related to the association of each sample to a class. The regression is then carried out between the X-block variables and the Y just established. This application for classification purposes is called PLS-DA. The conceptual difference between PCs and LVs is represented in Figure 5b: while PCs are aligned along the direction of maximum variance, LVs are aligned along the direction that maximizes the covariance between X and Y variables.

Figure 5: Calculation of a PLS model (a) and graphical representation of the differences between PCs and LVs (b).
PLS-DA and related procedures based on PLS are quite widespread in proteomics and several applications have been recently reported in clinical proteomics. West-Norager et al. [95] demonstrated the feasibility of serodiagnosis of ovarian cancer by MALDI-MS: 265 sera from women admitted with symptoms of a pelvic mass were used for model building. The authors developed a rigorous approach for building classification models suitable for highly multivariate data. Spectra were first aligned and zones not containing peaks were removed; finally a master list of 117 peaks defined by m/z intervals was obtained. For each interval, a PCA was calculated: if a peak represents only one underlying feature, it is expected that one PC can explain the variation; if the peak contains information from several chemical compounds, then more than one PC may be needed, but the number of PC will always be dramatically lower than the number of initial variables. Data redundancy was therefore eliminated by representing each peak by its relevant PCs. The entire procedure reduced the number of variables from more than 30000 to about 500. To test if further variable selection would improve model prediction, PLS-DA models were calculated using a stepwise variable selection based on using variable importance in projection (VIP) scores that estimate the importance of each variable used in the PLS-DA model: variables with VIP scores close to or greater than 1 were retained as significant. Time dependent changes in peak profiles up to 15 months after sampling were demonstrated, even when storing samples at -20°C. The best models were able to classify with 79% Spg and 56% Sg, i.e., an analytical accuracy of 68%.
Rajalahti et al. [96] applied PLS-DA to identify biomarker signatures in MS profiles of cerebrospinal fluid (CSF) from patients with multiple sclerosis. The low molecular weight CSF proteome from 54 patients with sclerosis and a range of other neurological diseases, as well as healthy controls, was analyzed in replicates using mass spectral profiling. PLS-DA identified the most discriminatory spectral regions by the exploitation of a nonparametric discriminating variable test (DIVA) together with the so-called selectivity ratio (SR) plot.
Finally, Yang et al. [58] identified prognostic polypeptide blood plasma biomarkers of Alzheimer’s disease (AD) progression. 119 blood plasma samples of patients with mild cognitive impairment (MCI) with different outcomes (stable and progressive MCI) were analyzed by untargeted, label-free shotgun proteomics. Predictive biomarkers of progressive MCI were selected by OPLS-DA [97], a modification of PLS developed for highly decorrelated datasets, which removes variation from X variables not correlated to Y: the final interpretation of the results is therefore easier due to the reduced complexity of the final model and usually the prediction ability of the model improves. The best model showed an accuracy of 79% in predicting progressive MCI. Some sex-specific protein biomarkers were also identified. Significant sex bias in AD-specific biomarkers underscores the necessity of selecting sex-balanced cohort in AD biomarker studies, or using sexspecific models.
Other applications regard: the proteomic profiling of follicular and papillary thyroid tumors [18]; the analysis of peptidomics data from clinical urine samples subjected to LC/MS to identify peptidebiomarker fingerprints related to disease diagnosis and progression [98]; the identification of a serum protein profile predictive of the resistance to neoadjuvant chemotherapy (NACT) in advanced breast cancers [56]; the identification of plasma protein biomarkers for depression and schizophrenia [99]; the identification of biomarkers in colon cancer by 2DE [17]. In this last application, PLS-DA was coupled to a variable selection procedure in backward elimination and compared to differential analysis carried out by classic PDQuest analysis: PLS-DA with backward elimination provided a larger set of candidate biomarkers proving to be more exhaustive than classic differential analysis.
LDA [79,80] is a Bayesian classification method providing the classification of the objects considering the multivariate structure of the data. In Bayesian methods, an object i, identified by its descriptor values xi, is assigned to class g for which the posterior probability P(g/ xi) is maximum.
Each class is usually described by a Gaussian multivariate probability distribution,
where the argument of the exponential function is the Mahalanobis distance between object i and the centroid of class g and takes into account the shape of the class and the correlations among the variables (it contains the covariance matrix). Each object is classified in class g if the so-called discriminant score Dg is minimum:
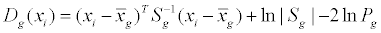
where Sg is the covariance matrix of class g, 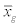 is the centroid of class g, Pg is the prior probability of class g.
is the centroid of class g, Pg is the prior probability of class g.
Bayesian methods differentiate according to how the covariance matrix is chosen: in LDA it is approximated with the pooled (between the classes) covariance matrix; this corresponds to consider all the classes as having a common shape (i.e. a weighted average of the shape of the classes present). Figure 6 shows an example of LDA where two classes A and B are present in the space described by two variables X1 and X2: while the two classes are overlapped along the two original variables, they appear separated along the discriminant function; Figure 6 reports the linear discriminant direction as a dotted line.

Figure 6: Example of LDA: the two classes A and B appear overlapped along both X1 and X2 directions, while they are perfectly separated along the discriminant function. The linear direction of separation between the classes is represented as solid line.
The variables contained in the LDA model discriminating the classes can be chosen by a stepwise algorithm, selecting iteratively the most discriminating variables. Usually, a Forward Selection (FS) procedure is applied: the method starts with a model where no variables are included and gradually adds a variable at a time until a determined criterion of arrest of the procedure is satisfied. The variable being included in the model in each step is the one providing the greatest value of an F-Fisher ratio, so that the j-th variable is included in the model, with p variables already included, if:
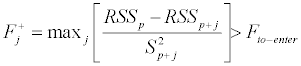
where:
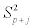 = variance calculated for the model with p variables plus j-th variable;
= variance calculated for the model with p variables plus j-th variable;
RSSp=residual sum of squares of the model with p variables;
RSSp+j=residual sum of squares of the model with p variables plus j-th variable.
The F value thus calculated is compared to a reference value (Fto-enter ) usually set at values ranging from 1 (more permissive selection, including a larger number of variables in the final model) to 4 (more severe selection).
LDA can be performed either on the original variables or on PCs. In this last case, it is possible to convert the LDA model based on the significant PCs in a model based on the original variables by means of the loadings. The combination of PCA and LDA allows the use of LDA even in cases when the data are characterized by fewer samples than variables, as it is usual in proteomics.
Another approach useful when the number of variables overcomes that of the samples is the use of DLDA, where the covariance matric is diagonal [100]. Since this simplification is by itself not enough when many variables are not relevant for the classification and they add noise, a Feature Subset Selection (FSS), which uses only a small fraction of the initial set of variables, can be used. Nearly all DLDA based techniques [100-102] use a filter approach for FSS: variables are first ranked using a statistical score and the discriminant function is built by selecting the highest ranking variables.
Many applications are present were LDA is applied alone or in combination with other methods like CART or PCA. An interesting theoretical paper was presented by Zollanvari et al. [103] who proposed the analytical formulation for the joint sampling distribution of the actual and estimated errors of a classification rule, applied to LDA. Error estimation must in facts be used to evaluate the accuracy of a designed classifier, an issue that is critical in biomarker discovery for disease diagnosis and prognosis in genomics and proteomics. Exact results are provided in the univariate case, and a simple method is suggested to obtain an accurate approximation in the multivariate case. The analysis presented is applicable to finite training data. In particular, it applies in the case of small-sample datasets commonly found in genomics and proteomics applications. Numerical examples illustrate the analysis.
For what regards the field of clinical chemistry, Imre et al. [28] applied LDA for the classification of cancer patients and controls based on the determination of N-glycans of human serum alpha-1- acid glycoprotein (AGP). N-glycan oligosaccharides of AGP samples isolated from 43 individuals (controls and patients with lymphoma and ovarian tumor) were analyzed by MALDI-TOF MS. 34 different glycan structures were identified. LDA analysis showed a good separation between the three groups (NER% 88%). Cross-validation results indicated that the method has predictive power: cancerous vs. controls showed 96% Sg and 93% Spg; lymphoma vs. controls + ovarian tumor cases instead 72% Sg and 84% Spg.
Another study regards the application of LDA in plant proteomics. LDA was coupled to PCA by Negri et al. [16] for the identification of proteins involved in biotic and abiotic stress responses in the ripening of Pinot Noir skins. A comparative 2-DE analysis of grape skins collected in three moments of ripening was carried out; PCA was applied to the spot volume dataset obtained and LDA with a variable selection procedure based on a forward stepwise search was applied to the obtained scores. This technique allowed to discriminate veraison, quite mature and mature samples, and to sort the matched spots according to their significance.
Other applications are by Zhong et al. [38] in animal proteomics and by Cheung et al. [104] in clinical proteomics (Table 3 for details).
This method is used to build classification rules. Classification trees [93] are built by subsequent divisions (splits) of subgroups of the original dataset in two descending subgroups with the aim of classifying the data in homogeneous groups as much as possible different one from the others. It is possible to derive a tree diagram where, starting from the root node (where the dataset is not separated), a series of nodes and branches separate; each node h represents a subgroup of the dataset. Nodes not undergoing a further split are called terminal nodes: to each terminal node a class is associated. Starting from the root node h1, the samples are separated in a series of splits: in each node the split giving the most homogeneous division of the data in the two descendent nodes is selected. An example is given in Figure 7.

Figure 7: Example of CART classification: objects belong to three classes (light grey=1, dark grey=2 and black=3). Nodes h3, h4, h5 are terminal nodes, while h1 and h2 represent splitting decisions. In h1, if the value of splitting variable 1 is > 1, the object is assigned to h2, otherwise to h3. In h2, if the value of splitting variable 2 is > 2, the object is assigned to h4, otherwise to h5.
Several applications of classification and regression trees are present in literature. One application is in the field of ecotoxicology [53] (Table 3 for details), while other applications are present in the field of clinical proteomics [35,36,39,54].
In the study by Whelan et al. [54], the authors applied LC-MS/MS to identify biosignatures of breast cancer in proximal fluid samples. The authors investigate three clinically important types of breast cancer using a panel of human cell lines: HER2 positive, hormone receptor positive and HER2 negative, and triple negative (HER2-, ER-, PR-). The most abundant secreted, sloughed, or leaked proteins released into serum free media from these breast cancer cell lines were characterized by a combination of protein fractionation methods before LC-MS/MS analysis. 249 proteins were detected in the proximal fluid of 7 breast cancer cell lines. Comparison of each cell line displayed unique and consistent biosignatures regardless of the individual group classifications, demonstrating the potential for stratification of breast cancer. Predictive CART was able to categorize each cell line as HER2 positive, HER2 negative and hormone receptor positive and triple negative based on only two proteins.
Other applications (Table 3 for more details) regard the search for biomarkers in: the response to Infliximab in Crohn’s disease [35]; hepatocellular carcinoma [39] and the predictive diagnosis of chronic allograft dysfunction by urinary proteomics [36]. All these applications regard the use of SELDI-TOF profiling.
Random Forests [105] is an extension of the classification trees and it is structured to grow many classification trees. The new objects are classified by each independent tree in the forest: each tree therefore gives a classification. The forest chooses the most recurrent classification (over all the trees in the forest). Each tree is grown as follows:
a. If N objects are in the training set, N cases are sampled randomly from the data to grow the tree;
b. If there are M variables, a number m<<M is specified such that at each node, m variables are selected at random out of the M and the best split on these m is used to split the node. The value of m is held constant during the forest growing.
c. Each tree is grown to the largest possible extent.
The error rate depends on: the correlation between any two trees in the forest (increasing the correlation increases the forest error rate); the strength of each individual tree in the forest (inversely correlated to the tree error rate).
RF are quite widespread for the identification of biomarkers in proteomics, the most of applications being in the field of clinical proteomics.
Ostroff et al. [106] investigated sera from 117 Malignant Pleural Mesothelioma (MM) cases and 142 asbestos-exposed control individuals for the early detection of MM. Biomarker discovery, verification, and validation were performed using SOMAmer proteomic technology, which simultaneously measures over 1000 proteins in unfractionated biologic samples. Using univariate and multivariate approaches 64 candidate protein biomarkers were identified; a 13-marker random forest classifier was derived with an AUC of 0.99 ± 0.01 in training, 0.98 ± 0.04 in independent blinded verification and 0.95 ± 0.04 in blinded validation studies. Sg and Spg were respectively 97% and 92% in training and 90% and 95% in blinded verification. This classifier accuracy was maintained in a second blinded validation set with a Sg/Spg of 90%/89% and combined accuracy of 92%.
Other applications (Table 3 for more details) regard: plasma proteomic profiles from disease-discordant monozygotic twins in multiple systemic autoimmune diseases by RP-LC-MS [57]; the investigation of how fasting for 36 h, as compared to 12h, affects the human proteome of platelets, peripheral blood mononuclear cells, plasma, urine and saliva [6]; the improvement of a multianalyte serum biomarker panel to identify lymphnode metastases in non-small cell lung cancer (NSCLC) with circulating autoantibody biomarkers [107]; the development of a multiplexed tumor-associated autoantibodybased blood test for detecting NSCLC [108] and the identification of biomarker panels in 2D-DIGE data from sera of patients with prostate cancer [19].
SVM [109] are machine learning algorithms that find a maximal margin hyperplane that maximizes the distance between the two classes present. A linear hyperplane is in general able to separate n samples in n+1 dimensions, therefore in high-dimensional data, the use of nonlinear kernels can be avoided. The linear SVM separates the classes by a linear boundary.
The graphical representation of a simple case is given in Figure 8: the solid line represents the border hyperplane, while the dotted lines delimit the border. Cortes and Vapnik [110] in 1995 proposed a modified maximum margin allowing misclassifications. Boser et al. [111] proposed also a modification to the original linear classifier providing a nonlinear classifier by applying the kernel trick to maximum-margin hyperplanes.

Figure 8: Example of SVM. The solid line represents the discriminant direction, while the dotted lines represent the margin boundaries.
Support vector machines have been extensively used in proteomics for the identification of biomarkers, usually coupled to other multivariate tools as genetic algorithms, PCA, random forests, Canonical Correlation Analysis (CCA). This last method is a multivariate generalization of the correlation analysis, developed by Hotelling; it is used to define the relationship between 2 sets of variables [80]. When CCA is applied to multichannel signal processing, linear combinations X and Y of two mean-centered multivariate random vectors [x1(t),...,xm(t)]T and [y1(t),...,yn(t)]T (t1,....,N) are defined.
CCA computes the linear combination coefficients (i.e. regression weights) ωx and ωy, so that the correlation between the new variables X and Y (called canonical variates) is maximum.
The first pair of canonical variates correspond to the eigenvectorsx and ωy associated with the largest eigenvalue. The remaining canonical variates correspond to the remaining eigenvectors, and the associated eigenvalues are the squared canonical coefficients. The canonical variates are maximally correlated and, at the same time, uncorrelated with the previous pairs.
Sparse Canonical Correlation Analysis (SCCA) is a modification of CCA [112] useful when CCA has to be applied to sparse data.
Karthikeyan et al. [113] applied SVM and the genetic algorithm (GA) to plasma peptide chromatograms for identifying biomarkers of air contaminant exposures. Interrogation of chromatographic data for biomarker discovery is hampered by the stochastic variability in retention times; the difficulty is further increased when the effects of exposure (e.g. to environmental contaminants) and biological variability result in varying numbers and intensities of peaks among chromatograms. The authors developed a software to correct the time shifts in chromatographic data through iterative selection of landmark peaks and isometric interpolation to improve alignment. To illustrate the tool, plasma peptides from Fischer rats exposed for 4h to clean air or Ottawa urban particles (EHC-93) were separated by HPLC with autofluorescence detection, and the retention time shifts between chromatograms were dewarped. Both dewarped and non-dewarped datasets were then mined for models containing peptide peaks that best discriminate among the treatment groups. In general, models generated by dewarped datasets were able to better classify test sample chromatograms into either clean air or EHC-93 exposure groups, and 0 or 24 h post-recovery time groups. Peak areas of peptides in a model that produced the best discrimination of treatment groups were analyzed by two-way ANOVA with exposure (clean air, EHC-93) and recovery time (0 h, 24 h) as factors. Statistically significant (p<0.05) time-dependent and exposure-dependent increases and decreases were noted establishing these as biomarker candidates for further validation.
Ahn et al. [114] identified serum biomarker panels for the diagnosis of gastric adenocarcinoma by random forests and SVM. A 29-plex array platform with 29 biomarkers, consisting of 11 proteins discovered through proteomics and 18 previously known to be cancerassociated, was constructed. Via RF, 13 markers were selected for multivariate classification analysis. Then, multivariate classification analysis with RF and SVM was performed on the training set, consisting of 70 serum samples from gastric adenocarcinoma patients and 70 control samples. Each algorithm was cross-validated on the test set to which 50 samples were assigned. The best RT and SVM algorithms showed mean accuracies of 88.3% and 89.7% respectively. These two algorithms were tested on a separate, independent blinded set of 95 gastric adenocarcinoma sera and 51 controls with mean accuracies of 89.2% and 85.6%, respectively. The biomarker panel selected by RF containing 11 markers showed a higher accuracy in the validation set. RF generally outperformed SVM, regardless of stage or tumour size even if SVM showed a higher sensitivity for small tumours.
Other applications (Table 3 for details) regard: the use of PCA and SVM to discriminate among multiple sclerosis-related disorders [30]; the comparative proteomic analysis of non-small-cell lung cancer and normal controls using serum label-free quantitative shotgun technology [59]; plasma biomarkers of tuberculosis and malaria by SCCA and SVM [112]; biomarkers associated with early renal injury by SELDI-TOF MS in human urine [37]; the ability to perform a clinical proteomic study using samples collected at different times from two independent clinical sites by label-free 2-D-LC-MS [51]; the development of a sequence-specific exopeptidase activity test for functional biomarker discovery [29].
In this section, some papers will be presented, reporting the use of alternative methods developed by different authors to address particular drawbacks of the standard multivariate tools.
Karimi et al. [115] proposed a novel approach for the identification of discriminatory variables in mass spectrometry by clustering of variables. In factor analysis-based discriminate models, latent variables are calculated from the data at all employed instrument channels. Since some channels are irrelevant for classification, the extracted LV’s possess mixed information from both useful and irrelevant channels. Clustering of variables (CLoVA) based on unsupervised pattern recognition was suggested but the authors as an efficient method to identify the most informative spectral regions. The m/z values were clustered into different clusters via self-organization maps. Then, the spectral data of each cluster were separately used as the input variables of classification methods such as PLS-DA and extended canonical variates analysis (ECVA). The proposed method was evaluated by the analysis of two experimental data sets (ovarian and prostate cancer datasets). The method was able to detect cancerous from control samples with higher Sg and Spg than conventional PLS-DA and ECVA.
Chen [27] applied multivariate curve resolution (MCR) to improve proteomic mass spectra classification. The paper describes a novel proteomic pattern analysis algorithm for biomarker discovery using MALDI-TOF or SELDI-TOF. The algorithm (MCR-marker) is based on the combination of MCR with classification methods and applies singular value decomposition to select differentially expressed m/z windows. In each selected m/z window, potential biomarkers are identified from MCR-resolved peak profiles that show better performance than the precise m/z values. The identified potential biomarkers are not dependent on the selection of MCR methods and consist of clearly detectable peaks, which may represent identifiable proteins, protein fragments or peptides. The algorithm was validated on two data sets from the literature.
Carlson et al. [116] reported the biomarker clustering to address correlations in proteomic data. Existing methods for dimension reduction, i.e. PCA and related techniques, are not always satisfactory in proteomics since they provide results that are of not easy interpretation. The authors propose a preprocessing algorithm that clusters highly correlated features, using the Bayes information criterion to select an optimal number of clusters. Statistical analysis of clusters, instead of individual features, benefits from lower noise, and reduces the difficulties associated with strongly correlated data. This preprocessing tool proved to improve biomarker discovery in clinical SELDI-TOFMS datasets of plasma from patients with Kawasaki disease and bone-marrow cell extracts from patients with acute myeloid or acute lymphoblastic leukemia.
Dutkowski and Gambin [117] proposed a new approach to the biomarker selection problem: the approach is based on the application of several competing feature ranking procedures and compute a consensus list of features based on their outcomes. The method was validated on two proteomic datasets for the diagnosis of ovarian and prostate cancer. The proposed methodology can improve the classification results and at the same time provide a unified biomarker list for further biological examinations and interpretation.
Han [118] applied nonnegative PCA for mass spectral serum profiles and biomarker discovery. Nonnegative PCA is an extension of PCA [118] where nonnegativity constraints are imposed to the loadings. This alternative is useful when PCA, providing both positive and negative loadings, makes the interpretation of the loadings quite difficult, i.e. when positive defined signals as spectra or mass signals are investigated. The author addresses the main drawback of PCA, i.e. its global feature selection mechanism that prevents it from capturing local features. In this study, the author developed a nonnegative PCA algorithm and present a nonnegative PCA based SVM algorithm with sparse coding to conduct a high-performance proteomic pattern classification.
Purohit et al. [41] developed a procedure for the identification of serum candidate biomarker of Type 1 diabetes (T1D) by SELDI-TOF and model averaging. 146 protein/peptide peaks were identified as significantly changing over a total of 581 peaks discovered. The data were split for the first replicate into training and test sets. Normal kernel discriminant analysis was then used to obtain random sets of three peaks (model), and each of 200000 models of three peaks was evaluated using LOO cross-validation. Models with LOO crossvalidation error rates >25% were discarded, and the predictions of the remaining models were averaged using plurality voting. The resulting set of models was then evaluated on the test set. The identified models were then applied to the dataset for the second replicate. T1D and control samples were classified with 88.9% Spg and 90.0% Sg, while 82.8% Spg and 76.2% Sg were reached on the test set.
Other applications (Table 3 for details) regard: a protein biomarker profile in tear fluid for breast cancer patients by artificial neural networks on SELDI-TOF MS [42]; a comparative urine analysis by liquid chromatography-mass spectrometry and PCA coupled to the Nearest Shrunken Centroid (NSC) algorithm [46]; the correlation of GC-TOFMS- based metabolite profiling and LC-MS-based protein profiling to improve pattern recognition for multiple biomarker selection, by PCA and Independent Component Analysis (ICA) [47]; the extraction of reliable protein signal profiles from MALDI-TOF spectra by ICA [24]; the identification of a liver cirrhosis signature in plasma for predicting hepatocellular carcinoma risk by PCA, DLDA and 3-Nearest Neighbors (3-NN) [119]; a high performance profile-biomarker diagnosis for mass spectral profiles by embedding multi-resolution ICA in LDA and SVM [120]; the application of multivariate adaptive regression splines analysis to predict biomarkers of spontaneous preterm birth [121]; the urine proteomic profiling of pediatric nephrotic syndrome by a genetic algorithm search in the principal component space [40].
For what regards the development of software tools for multivariate data treatment, Fan et al. [122] developed digger, a graphical user interface R package for analyzing 2D-DIGE data by different multivariate tools. Akella et al. [48] instead developed CLUE-TIPS (Clustering Using Euclidean distance in Tanimoto Inter-Point Space), a clustering method for pattern analysis of LC-MS Data. In CLUETIPS, an intersample distance feature map is generated from filtered, aligned and binarized raw LC-MS data by applying the Tanimoto distance metric to obtain normalized similarity scores between all sample pairs for each m/z value. Clustering and visualization methods for the intersample distance map were developed to analyze datasets for differences at the sample level as well as the individual m/z level. CLUE-TIPS can also be used as a tool in assessing the quality of LCMS runs. It was applied to LC-MS data obtained from plasma samples collected at various time points and treatment conditions from immunosuppressed mice implanted with MCF-7 human breast cancer cells. CLUE-TIPS successfully detected the differences/similarities in samples at various time points taken during the progression of tumor, and also recognized differences/similarities in samples representing various treatment conditions.
Some interesting papers have recently appeared focused on the comparison of different multivariate methods: these studies are reported here together with the main findings to provide information on the performance of different statistical tools when they are applied to the same case study.
Brasier et al. [123] examined physiological data from 1048 subjects to identify 4 quantitative intermediate phenotypes asthma. Four different statistical machine learning methods were evaluated to predict each intermediate phenotype using cytokine measurements on a 76 subject subset. The comparison of these models using the area under the ROC curve and the overall classification accuracy indicated that logistic regression and multivariate adaptive regression splines produced the most accurate methods to predict intermediate asthma phenotypes.
Levner [124] reported the application of feature selection and NSC classification for protein mass spectrometry; the aim of the study was to reduce data dimensionality in mass spectrometry to allow the use of standard machine learning techniques. The performance of the NSC classifier was evaluated coupled with different feature selection algorithms: Student-t test, Kolmogorov-Smirnov test, P-test, sequential forward selection and a modified version of sequential backward selection. In addition, several dimensionality reduction approaches were tested: PCA and PCA coupled with LDA. Comprehensive experiments, conducted on five popular cancer datasets, revealed that the sequential forward selection and boosted feature selection algorithms produced the most consistent results across all data sets.
Guo and Balasubramanian [125] performed the comparative evaluation of classifiers in the presence of statistical interactions between features in high dimensional data settings. A central challenge in biomedical investigations involves the estimation of an optimal prediction algorithm to distinguish between different disease phenotypes: these analyses are hampered by features that exhibit statistical interactions. The authors compared the performance of 4 classifiers (K-NN, Prediction Analysis for Microarrays - PAM, RF and SVM) in settings involving high dimensional datasets including statistically interacting feature subsets. Their performance was evaluated under varying sample size, levels of S/N ratio and strength of statistical interactions among features. Simulation studies revealed that the classifier PAM had the highest classification accuracy in the absence of noise, statistical interactions and when feature distributions were multivariate gaussian within each class. In the presence of statistical interactions, modest effect sizes and the absence of noise, SVM achieved the best performance followed closely by RF. RF was optimal in settings that included both significant levels of high dimensional noise features and statistical interactions between biomarker pairs.
Christin et al. [126] compared different feature selection methods for biomarker discovery in clinical proteomics. Six feature selection methods for LC-MS-based proteomics and metabolomics biomarker discovery were compared: t test, Mann-Whitney-Wilcoxon test (MWW test), NSC, linear SVM-recursive features elimination (SVM-RFE), PCA-DA and PLS-DA. The methods were tested using human urine and porcine cerebrospinal fluid samples that were spiked with a range of peptides at different concentration levels. The ideal feature selection method should select the complete list of discriminating features that are related to the spiked peptides without selecting unrelated features. The performance was assessed using the harmonic mean of the recall and the precision (f-score) and the geometric mean of the recall and the true negative rate (g-score). The univariate t and MWW tests with multiple testing corrections are not applicable to data sets with small sample sizes (n=6), but their performance improves markedly with increasing sample size up to a point (n>12) at which they outperform the other methods. PCA-DA and PLS-DA select small feature sets with high precision but miss many true positive features related to the spiked peptides. NSC strikes a reasonable compromise between recall and precision for all data sets independent of spiking level and number of samples. Linear SVM-RFE performs poorly for selecting features related to the spiked compounds, even though the classification error is relatively low.
Ongay et al. [127] performed the statistical evaluation of CZE-UV and CZE-ESI-MS data of intact alpha-1-acid glycoprotein isoforms for their use as potential biomarkers in bladder cancer. Samples from 16 individuals (8 healthy, 8 bladder cancer) were analyzed. The analytical data were evaluated employing different statistical techniques: ANOVA, PCA, LDA and PLS-DA. Statistically significant differences between the two groups of study were observed. The best results were obtained by LDA that showed a NER=93.75%.
This review is aimed to present the most recent applications of multivariate statistical tools in proteomics for the identification of biomarkers. The most recent applications present in literature were presented separately for the different multivariate methods adopted together to the theoretical bases of each statistical method. A quite wide range of different statistical methods are exploited in literature for the identification of biomarkers in proteomics, providing sound results. In general, multivariate methods should always be preferred to univariate approaches to provide a pool of markers highlighting synergistic and antagonistic effects. The biological effect played by a particular factor (a pathology, a drug, a polluting effect, a ripening effect etc) is usually the result of a series of different mechanisms either independent from each other or showing relevant interactions. Multivariate procedures are able to highlight these relationships avoiding the neglection of relevant information.
It is however important to avoid the identification of false positives, mostly due to chance correlations: this risk greatly increases when few information is available (i.e. a small number of cases investigated), as is often the case in proteomic studies. This problem can be partially solved by a sound experimental design and sample collection; each study should be carefully designed from a statistical point of view before being performed in order to include all possible sources of biological variation. In the cases when the poor availability of samples cannot be solved, it is very important to apply mathematical tools to validate the models built, thus evaluating the predictive ability of the models: in these cases cross-validation or the use of simulation algorithms is mandatory to identify only statistically significant markers. Another way to face this problem is the use of multivatiate methods based on projection to latent structures (e.g. based on PCA and PLS approaches), able to provide a substantial dimensionality reduction by considering few latent variables or principal components rather than a large number of original variables. This approach makes also possible the application of other classification tools as LDA to problems where a smaller number of samples than of variables is present: in such cases in fact LDA cannot be applied unless a variable selection procedure is applied providing a maximum number of discriminant variables equal to the number of samples available.
The identification of biomarkers represents a balance between the achievement of parsimonious models with the best predictive ability and the necessity of obtaining the maximum amount of information about the effect investigated: it is the authors’ opinion that the future perspective in biomarkers identification has to be searched for in the exhaustive search for potential markers. Complex effects as pathologies, pollution effects, drug effects etc, acting on a wide range of individuals characterized by a large biological variability cannot in fact realistically reflect in a restricted panel of biomarkers. We think that the future will rely on high-throughput techniques that are able to provide great amount of information that can be coupled together to identify exhaustive panels of markers, improving the predictive performance of the final models in terms of specificity and sensitivity: to this respect the use of sound and reliable multivariate tools is particularly important to obtain reliable results.