Pancreatic Disorders & Therapy
Open Access
ISSN: 2165-7092
ISSN: 2165-7092
Research Article - (2015) Volume 5, Issue 2
Collecting repeat samples of blood ("liquid biopsies") is a broadly used clinical approach for serial monitoring of disease or response to treatments. In patients with cancer the most distinct molecular feature are somatic mutations acquired by cancer cells present in the diseased tissue. Indeed, mutant DNA derived from dying or lysed cancer cells can be isolated from patient serum samples, subjected to DNA sequencing and to analysis of abundance as a measure of tumor burden. Also, changes in the DNA mutation patterns in serum samples collected over time can indicate altered pathways or clonal evolution of the disease and altered abundance of mutant DNA suggests an altered disease burden. In addition, during the course of treatment, changes in circulating DNA mutation patterns can indicate the emergence of resistant clones and prompt changes in treatment. In contrast to mutant DNA, microRNAs (miR) are transcribed, processed, packaged and released from cells in normal and in diseased tissues as part of the extracellular crosstalk between cells. Interestingly, released miR can function in cell-to-cell communication and as hormone-like signals that operate at a distance through their release into the circulation and subsequent uptake into cells in distant tissues. Circulating miR expression patterns can be established from serial serum samples and monitored for alterations over time. Circulating miR provide a readout of the organism's steady state and serial analyses will indicate changes in the response to therapy or an altered physiologic or disease state. Furthermore, changes in circulating miR patterns can indicate treatment efficacy or resistance as well as adverse effects associated with the respective intervention. Thus, the combined serial analysis of mutant DNA and miR in the circulation has the potential to provide a molecular footprint of pancreatic cancer and can be used to monitor treatment responses or resistance to treatment in real time with a minimally invasive procedure.
<Keywords: Pancreatic cancer, Liquid biopsy, Biomarkers, Circulating DNA, Circulating microRNA, Prognostic markers, Treatment response
Pancreatic cancer is a deadly malignant disease with surgical resection currently the only potentially curative modality although only 10-20% of patients are diagnosed with a resectable tumor [1,2]. Patients with advanced or metastatic disease receive chemotherapy with either gemcitabine or S-1, an oral fluoropyrimidine, or with the combination treatment FOLFIRINOX [3-5]. The prognosis of patients receiving surgery and chemotherapy is dependent on a number of characteristics including radiographical staging, histological grading, ECOG scores and CA19-9 levels [6-8]. However, very little improvement in the outcomes of patients with pancreatic cancer has been seen in the past 30 years [1]. Here we will review the potential to use blood samples or "liquid biopsies", a recent term coined to indicate the relation to cancer tissue analysis, as markers of genetic characterization of a cancerous lesion. It is conceivable that blood samples can reflect the mutation profile of a primary cancer as well as residual cancer cells that are not accessible physically or below detection. Additionally, liquid biopsies can be taking repeatedly to follow evolution of the disease. Identification of specific pancreatic cancer markers is crucial for the improvement of controlling this disease and recent studies have shown that circulating mutant DNA can be found in the majority of cancer patients and can provide a measurement of tumor burden. Moreover, it could be used to identify specific mutant genotypes emerging from a heterogeneous tumor population. In contrast to the unique source of mutant DNA, i.e. cancer cells, miR isolated from the circulation is derived from virtually all tissues in the organism (Figure 1.) The analysis of expression patterns of circulating miR will thus provide a footprint of the steady state of the organism that encompasses normal as well as pathologic processes. Also, it will reflect the impact of therapeutic interventions on the diseased as well as healthy tissues. Here we review the published data on circulating nucleic acids, i.e. DNA and miR that reflect the disease state and treatment responses of patients with pancreatic cancer.
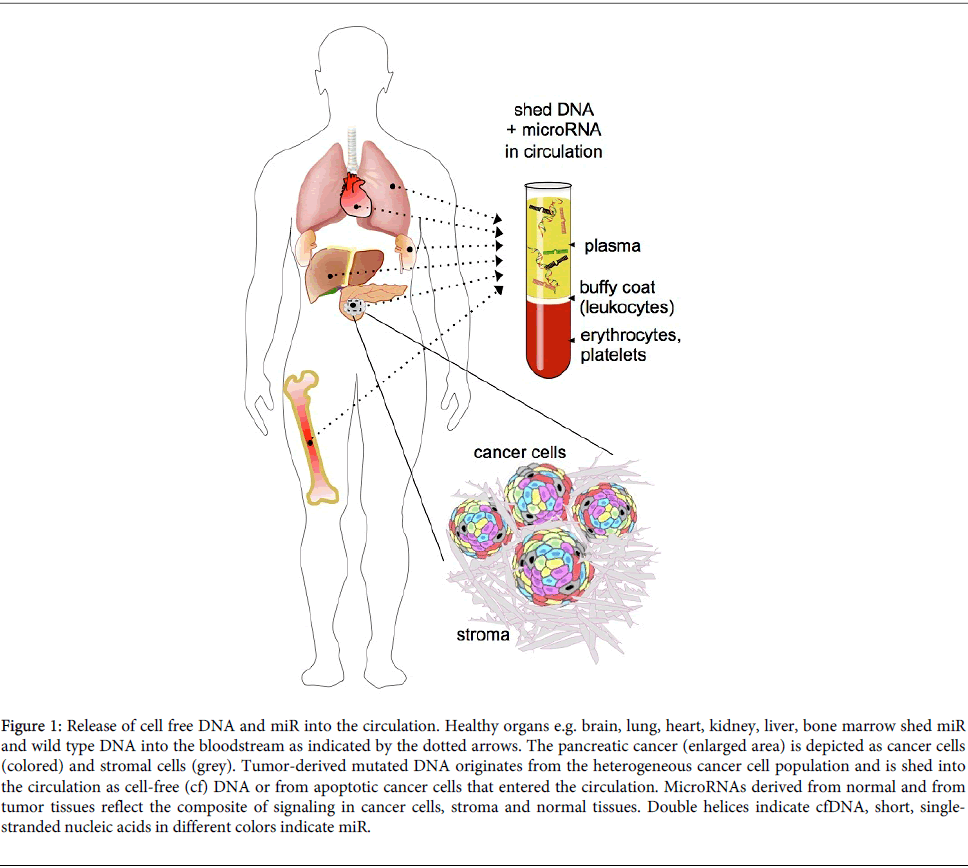
Figure 1: Release of cell free DNA and miR into the circulation. Healthy organs e.g. brain, lung, heart, kidney, liver, bone marrow shed miR and wild type DNA into the bloodstream as indicated by the dotted arrows. The pancreatic cancer (enlarged area) is depicted as cancer cells (colored) and stromal cells (grey). Tumor-derived mutated DNA originates from the heterogeneous cancer cell population and is shed into the circulation as cell-free (cf) DNA or from apoptotic cancer cells that entered the circulation. MicroRNAs derived from normal and from tumor tissues reflect the composite of signaling in cancer cells, stroma and normal tissues. Double helices indicate cfDNA, short, singlestranded nucleic acids in different colors indicate miR.
The occurrence of cell-free, circulating DNA (cfDNA) in the blood of patients was first described several decades ago [9-12]. Whilst the concentration of cfDNA in the bloodstream of healthy individuals is very low, cfDNA abundance increases significantly due to pathological processes, as reviewed by Anker et al. [13]. Elevated levels of cfDNA are detected in patients after surgery and trauma [14], myocardial infarction [15-17], rheumatoid arthritis [9], pancreatitis [18] and intensive care related conditions [19,20]. Additionally, athletes have increased levels of cfDNA after heavy exercise [21] and pregnant women carry fetal DNA in their bloodstream that has been used in prenatal diagnostics to detect fetal abnormalities [22-24].
Despite the multitude of studies describing cfDNA, the origin and nature of the double-stranded nucleic acid fragments isolated from the circulation remain ambiguous [25,26]. It is commonly accepted that apoptotic cells release DNA into the bloodstream. During apoptosis DNA is cleaved and packaged into nucleosomes of roughly 180 base pairs [27,28] and analysis of the size of circulating DNA indicates that at least some is from debris after the apoptotic process [29-32]. Additionally, there is some evidence of lymphocytes and cancer cells shedding DNA by unknown mechanisms [25,32-35]. Even though the complex origin of cfDNA needs to be further elucidated, elevated levels of cfDNA have been shown in independent studies of cancer patients when compared to healthy individuals. This elevated cfDNA is indeed mostly derived from cancer cells [10,11,30,36-39].
It is widely accepted that circulating mutant DNA originates from cancer cells. However as studies accumulate, it becomes clear that a multitude of aspects contribute to the varying levels of mutant DNA. Firstly, biological processes influence the overall quantity of circulating DNA. Circulating mutant KRAS DNA can be excreted by the kidneys and can be detected in the urine [40]. Plasma nucleases [41] and DNase1 [42] can contribute to the degradation of circulating DNA. Furthermore, DNA can be bound to proteins [43,44] and leukocyte activity influences the levels of cfDNA [42]. Intra-tumor heterogeneity and the amount of wild type tumor stroma DNA will have a great impact on the relative amount of mutant cfDNA [45]. Furthermore, age impacts the amount of cfDNA: Individuals of >90 years of age have elevated levels of cfDNA. Also, fragmented patterns and appearance of low molecular weight cfDNA was observed in the majority of the nonagenarians [46].
Secondly, technical aspects must be taken into account when assessing the quantity and quality of cfDNA. Levels of cfDNA are higher in plasma samples compared to serum samples because EDTA indirectly inhibits blood DNases [47]. Blood sample storage before spinning affects both the amounts and integrity of cfDNA [48] due to ongoing lysis of leukocytes before they are separated from the plasma by centrifugation. Furthermore, the wide variety in mutation detection techniques will impact sensitivity and specificity [38,49,50]. More recent studies report the use of targeted amplicon sequencing or massive parallel sequencing to cover a range of genes with great depth [51,52]. By using these recent approaches, the genomic heterogeneity as well as tumor burden can be revealed in more detail and with greater reliability than was possible even a few years ago.
Genomic mutations in the Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS) gene are found in 80 to 90% of all pancreatic adenocarcinomas [53-55]. As a consequence, cfDNA from cancer patients should harbor these mutations and will be indicative of tumor-derived DNA. Data from numerous studies show the presence of mutant KRAS DNA in the circulation of patients with pancreatic cancer and are summarized in (Table 1). These results from a number of research groups show that the detection of circulating mutant KRAS DNA can be used to monitor pancreatic cancer progression. The diversity in PCR and sequencing techniques used across the studies, however, contributes to a varying detection rate of circulating mutant KRAS DNA. Also, the various technical aspects regarding cell free DNA isolation influence the quality of the mutant DNA measurements as discussed above. With respect to the specificity of mutant KRAS cfDNA for the detection of pancreatic cancer, the findings are mixed. Four studies did not detect circulating mutant KRAS DNA in patients with benign pancreatic pathology.
| number of patients with pancreatic cancer patients with circulating mutant KRAS DNA |
detection method | pancreatic cancer stage | number of patients without pancreatic cancer with circulating mutant KRAS DNA remarks |
remarks | reference |
| 17 / 21 (81%) | PCR-RFLP followed by sequencing | unresectable disease |
0 / 5 healthy 0 / 3 CP |
2 patients had circulating mutant KRAS without detected mutant KRAS in the cancer tissue |
Mulcahy et al. 1998 [109] |
| 29 / 41 (71%) | PCR-RFLP followed by gel electrophoresis |
stages I-IV | 0 / 21 healthy 0 / 4 CP 0 / 3 cyst adenoma |
no relationship with tumor stage |
Dainxu et al. 2002 [110] |
| 9 / 15 (60%) | MASA-PCR followed by gel electrophoresis |
stages I-IV | 0 / 5 healthy 0 / 4 CP |
related to tumor size / persistant circulating mutant KRAS DNA 3 out of 15 patients after surgery and/or chemoradiation |
Yamada et al. 1998 [58] |
| 22 / 47 (47%) | allele-specific PCR followed by gel electrophoresis |
stages I-IV | 4 / 31 CP | patients with CP did not get pancreatic cancer in 36 months follow up time / no association with tumor stage or survival |
Maire et al. 2002 [56] |
| 20 / 56 (36%) | RT-PCR using PNA clamping |
operable + unoperable disease |
0 / 13 CP | no association with tumor resectability or survival |
Dabritz et al. 2009 [111] |
| 9 / 26 (35%) | mismatch ligation assay |
stages I-IV | - | persistant circulating mutant KRAS DNA after surgery in 4 out of 9 patients / no association with tumor stage, grade or metastasis |
Uemura et al. 2004 [112] |
| 30 / 91 (33%) | PCR followed by sequencing |
unresectable disease |
- | related to stage, metastasis and survival |
Chen et al. 2010 [59] |
| 12 / 44 (27%) | PCR-RFLP followed by gel electrophoresis |
stages I-IV | 0 / 4 healthy 2 / 37 CP |
no association with tumor size, however related to stage, metastasis and survival |
Castells et al. 1999 [57] |
| (RT) PCR = (real time) polymerase chain reaction CP = chronic pancreatitis RPFL = restriction fragment length polymorphism MASA = microsatellite associated sequence amplification PNA = peptide nucleic acid |
|||||
Table 1: Summary of published reports on circulating mutant KRAS DNA in patients with pancreatic cancer.
However, two studies described mutant KRAS in the circulation of 13% and 5% of patients (4 of 31 [56]; 2 of 37 [57]) with chronic pancreatitis. These patients then underwent surgery to remove the diseased organ. However, surgical pathology analysis of the tissues did not detect pancreatic cancer and after a follow-up of 6 to 36 months, none of the chronic pancreatitis patients with cfDNA KRAS mutations had developed pancreatic cancer. The transiently detected mutant KRAS DNA in the circulation likely originated from hyperplastic, mucous cells of chronically inflamed pancreas that had been removed and it thus appears that mutant KRAS cfDNA is not a reliable indicator of the presence of pancreatic cancer.
The majority of published studies on pancreatic cancer utilize mutant KRAS cfDNA as a diagnostic marker to confirm the presence of malignancy. When assessing alterations in circulating mutant KRAS as a marker of altered tumor size, one group found no correlation [57], whilst another group did find tumor size and abundance of circulating mutant KRAS cfDNA to be correlated [58]. Overall, the ratio of cancer cell to desmoplastic wildtype stroma will likely impact the abundance of circulating mutant KRAS and variability in the assessment of tumor / stroma ratio could explain the inconsistent findings across studies.
Furthermore, the abundance of mutant KRAS cfDNA could serve as a prognostic marker. Studies by Castells et al. and Chen et al. conclude that the abundance of circulating mutant KRAS DNA is associated with disease stage, metastasis and survival [57,59]. Uemura et al. found circulating mutant KRAS DNA in 5 out of 25 patients with stage III pancreatic cancer, in contrast to 25 out of 37 patients with stage IV disease [112]. Importantly, they showed that the survival of 30 patients with detectable KRAS mutations in circulating DNA is 6.3 months shorter than that of patients where only wildtype KRAS DNA was detected (p<0.001). After surgical removal of the tumor, cfDNA can serve as an indicator for the presence of residual cancer cells. Uemura et al. [112] showed that 5 out of 9 surgeries resulted in the disappearance of mutant KRAS cfDNA. Additionally, Yamada et al. [58] found that surgery and/or chemoradiation treatment resulted in the disappearance of mutant KRAS from plasma DNA in 12 out of 15 patients. A recent study by Newman et al. [38] in lung cancer patients they used a more sensitive DNA detection method for mutant cfDNA levels and found a correlation with tumor volumes during chemotherapy and surgery and with CT and PET scan data. Thus, technical aspects of sample handling, tumor size measurements and DNA measurement may account for the inconsistencies seen in the earlier pancreatic cancer studies.
In contrast to blood samples, small tissue biopsies will provide limited and possibly erroneous genetic characterization due to the spatial and temporal intra-tumor heterogeneity [60]. The mutation pattern in a tumor can be indeed derived from deep sequencing analysis of cfDNA [51,52] and cfDNA can be used to reveal clonal evolution due to drug treatments [51,61]. During the course of treatment, the somatic alterations of resistant clones can be accurately followed using cfDNA, which can be applied to tailor the therapy to a specific patient. No such studies have been published in pancreatic cancers. A study by Diaz et al. on mutant KRAS cfDNA in colon cancer patients could serve as a paradigm [62]: They found that 9 of 24 patients whose colorectal tumors were initially KRAS wildtype, developed detectable mutations in KRAS in their cfDNA after treatment with an EGFR antibody. The appearance of these mutations was very consistent, occurring between 5 and 6 months following treatment. This suggests that the emergence of KRAS mutant clones was due to the selection for resistance to EGFR blockade. These findings suggest potential utility also for pancreatic cancer monitoring via cfDNA analysis.
MiR are non-coding ribonucleic acids of ~22 nucleotides in length, which regulate mRNA stability and translation of known protein coding genes [63]. MiR genes are transcribed from individual genetic loci [64] and show complementary binding to 3' or 5'-untranslated regions (UTR) of the targeted mRNA or to the open reading frames. Via this interaction miR can lead to mRNA degradation or inhibition of protein translation [65,66]. Due to the relatively short seed sequence a single miR may target hundreds of mRNAs. Thus miR are involved in virtually all physiological and pathological processes and found altered in every type of cancer studied to date [67].
Cells export miR -containing apoptotic bodies, shedding vesicles, and exosomes into the bloodstream [68-71]. Microvesicles are impermeable to RNases, which explains the remarkable stability of extracellular miR. Additionally, a majority of extracellular miR in plasma or serum are membrane vesicle-free, but associated with 1 of 4 proteins of the Argonaute (AGO) family [72]. The remarkable stability of AGO2 protein explains the stability of associated miR even in nuclease- and protease-rich environments [71]. Data from a number of laboratories suggest that different RNA species can be specifically packaged into microvesicles by active sorting mechanisms which have not been fully elucidated [73]. Blood cells are major contributors to extracellular miR content in the circulation [74]. Different organs contribute to circulating miR because tissue-specific miR such as miR -122 (liver), miR -133a (muscle), miR -208a (heart), and miR - 124 (brain) have been consistently detected in plasma samples [75-77]. Tumors release miR into the bloodstream and cancer tissue-specific miR have been found in the circulation at different stages of the disease (for a recent review of circulating miR in cancer see Schwarzenbach et al. [78]).
Circulating miR entrapped within microvesicles can be transferred to recipient cells as signaling molecules and alter gene expression in target cells [73]. Once in the circulation, miR can function as hormones and a given miR may function as an oncogene or a tumor-suppressor gene depending on the cellular context and the targeted organ [79]. The concentration of miR in the bloodstream reflects alterations in the homeostasis of the entire organism and a panel of miR can easily be quantitated by quantitative PCR [80]. Findings from such studies have established the basis for circulating miR as disease biomarkers.
Cell free circulating miR can be obtained by using plasma that is collected in EDTA tubes or serum collected in the absence of anti-coagulants. Both can produce satisfactory results [81]. However, plasma collection using heparin as an anticoagulant interferes with the quantitation of miR species using PCR-based approaches [82]. Even though this inhibition can be alleviated by digesting heparin with lyase heparinase I prior to reverse transcription, it leads to lower efficiency and higher costs. Blood samples have to be centrifuged promptly to separate the cell-free phase and prevent hemolysis. Excessive concentrations of miR-16 and miR-451 can be indicative of hemolysis [83]. Before extraction of circulating miR, cells must be removed by centrifugation at about 1,200 x g. Furthermore, dead cells, apoptotic bodies, large shed vesicles, and cell debris can be removed by high-speed centrifugation 14,000 to 16,000 x g [84]. To monitor the efficiency of miR isolation, synthetic miR controls (SYNTH) can be used by spiking samples. When extracting miRs from blood samples, endogenous inhibitors of polymerases must be removed as much as possible: Trizol reagents are an example of reagents that will serve this purpose [85]. Polymerase inhibitors may include IgGs, hemoglobin or lactoferrin. Measuring miR levels by Taqman qPCR has become a standard method. In the case of very low RNA yields, the use of Locked Nucleic Acid-enhanced primers improves the sensitivity of the qPCR [86]. Furthermore, mutated Taq polymerases are less sensitive to blood-borne inhibitors. A Taq polymerase mixture of normal and Hemo KlenTaq improves sensitivity up to 30-fold as reported by Kim et al. [81]. To account for different extraction efficiency, different approaches have been used to compare miR concentrations across samples. If a large number of miR is detected in a single sample, the mean (or median) abundance can be used for normalization. There are many reports on using levels of endogenous miRs as internal reference control [87-89]. However, caution has to be taken before using this approach, since biological and technical processes can greatly influence the abundance of the miRs.
A wealth of information indicating the potential use of circulating miR for pancreatic cancer screening has emerged over the past years (Table 2). In 2014, Ganepola et al. performed a non-biased screening method to develop a panel of blood based diagnostic biomarkers consisting of circulating miR for the detection of pancreatic cancer at an early stage. They compared 8 early stage pancreatic cancer patients to 11 healthy controls and performed a high throughput screening using hybridization microarray analysis [90]. After validation studies, 3 miR, miR -22, miR -642b and miR-885-5p were confirmed to show consistent expression between microarray and qPCR. These 3 miR were then validated and evaluated as a diagnostic panel with a new cohort of patients and controls and found to yield 91% sensitivity and 91% specificity. The CA19-9 marker showed 73% sensitivity and 100% specificity.
| Pancreatic cancer vs. benign pancreas pathology (n) | Circulating miRs | Pancreatic cancer vs. healthy pancreas (n) | Circulating miRs | Remarks | Reference |
| - | - | 28 vs. 19 | miR-21, miR-210, miR-155, miR-196a (all ↑) |
- | Wang et al. 2009 [113] |
| - | - | 28 vs. 19 | miR-200a, miR-200b (both ↑) |
benign pathology vs. healthy miR-200a, miR-200b (both ↑) |
Li et al. 2010 [17] |
| - | - | 50 vs. 10 | miR-21, (↑) miRlet- 7d, miR-146a (both ↓) |
prognostic, survival | Ali et al. 2010 [101] |
| - | - | 22 vs. 25 | miR-210 (↑) |
no correlation with CA19-9 levels | Ho et al. 2010. [114] |
| 6 vs. 8 | miR-100a, miR-10 (both é) |
- | - | miR-10 (↑)homology with pancreatic cancer-bearing mice / miR-155 (↓) tumor specific gemcitabine response in pancreatic cancer-bearing mice |
LaConti et al. 2011 [107] |
| - | - | 36 v. 30 | miR-18 (↑) | miR-18 (↑) after surgery | Morimura et al. 2011 [104] |
| 140 vs. 111 | miR-21, miR-155, miR-16, miR-181a, miR-181b, miR-196a (all ↑) |
140 vs. 68 | miR-21, miR-155, miR-16, miR-181a, miR-181b, miR-196a (all ↑) |
benign pathology vs. healthy: miR-155, miR-181a, miR-181b, miR-196A (all ↑) |
Liu et al. 2012 [93] |
| 41 vs. 35 | miR-24, miR-134, miR-146a, miR-378, miR-484, miR-628-3p, miR-1290, miR-1825 (all ↑) |
41 vs. 19 | miR-24, miR-134, miR-146a, miR-378, miR-484, miR-628-3p, miR-1290, miR-1825 (all ↑) |
miR-1290 and miR-486-3p prognostic |
Li et al. 2013 [105] |
| 109 vs. 38 | miR-182 (é) | 109 vs. 50 | miR-182 (é) | prognostic | Chen et al. 2014 [115] |
| 77 vs. 67 | miR-10b, miR-30c, miR-106b, miR-132, miR-155, miR-181a, miR-181b, miR-196a, miR-212 (all ↑) |
77 vs. 71 | miR-10b, miR-30c, miR-106b, miR-132, miR-155, miR-181a, miR-181b, miR-196a, miR-212 (all ↑) |
benign pathology vs. healthy miR-212, miR-155 (both ↑) |
Cote et al. 2014 [92] |
| - | - | 11 vs. 11 | miR-642b, miR-885-5p, miR-22 (all ↑) |
- | Ganepola et al. 2014 [90] |
| - | - | 49 vs. 27 | miR-492, miR-663a (both ↓) |
- | Lin et al. 2014 [116] |
| 32 vs. 12 | miR-483-3p (↑) | 32 vs. 30 | miR-21, miR-483-3p (both ↑) |
miR-21 prognostic | Abue et al. 2015 [102] |
| 198 vs. 21 | miR-6826-5p, miR-6757-5p, miR-3131, miR-1343-3p (all ↑) |
100 vs. 150 | miR-6075, miR-4294, miR-6880-5p, miR-6799-5p, miR-125a-3p, miR-4530, miR-6836-3p, miR-4634, miR-7114-5p, miR-4476 (all ↓) |
pancreato-biliary cancer taken together |
Kojima et al. 2015 [91] |
Table 2: Summary of published reports on circulating miRs in patients with pancreatic cancer and benign pancreatic pathology.
In a large study by Kojima et al. 571 serum samples obtained from healthy subjects, patients with pancreatic, biliary-tract, or other digestive cancers and patients with non-malignant abnormalities in the pancreas or biliary tract were analyzed. Expression levels of miRs were obtained by a 3D-Gene microarray. They found that a combination of 8 miRs (miR-125a-3p, miR-4294, miR-4476, miR-4530, miR-6075, miR-6799-5p, miR-6836-3p and miR-6880-5p) achieved the best discriminant performance. The sensitivity was 80.3%, specificity 97.6% and accuracy 91.6% in detecting pancreatobiliary cancers contrasted with healthy controls, non-malignant abnormalities or other types of cancers [91].
There are some reports that extend beyond cancer related miRs, which show that similar miRs are indicators of benign pancreas pathology. Chronic pancreatitis is a risk factor for pancreatic cancer and shares clinical symptoms with pancreatic cancer. For example, circulating miR-155, 181a, 181b, 196a, 200a, 200b, and 212 were reported as significantly upregulated in patients with chronic pancreatitis versus healthy controls [17,92-94]. Li et al. showed that levels of serum miR-200a and miR-200b are similar in pancreatic cancer and pancreatitis patients, although they are increased compared to healthy controls [94]. Interestingly the miR-200 family can inhibit epithelial to mesenchymal transition and may thus have a direct signaling role [95]. In conclusion, circulating miRs can indicate the presence of chronic inflammatory and potentially premalignant processes in the pancreas.
Pancreatic cancer is a systemic disease that encompasses altered glucose metabolism [96,97], stroma remodeling [98], and bone marrow derived cellular activity [99,100]. The involvement of circulating miRs in such processes makes them excellent candidates to reflect the systemic impact of the disease and may provide additional molecular markers derived from other tissues impacted by the disease, e.g. the bone marrow. Ali et al. found that a high level of plasma miR-21 was correlated with worse survival, and the expression of let-7 was inversely correlated with survival [101]. In addition, Abue et al. tested the levels of miR--‐483-3p and miR-21 in plasma of 32 pancreatic cancer patients, 12 patients with intraductal papillary mucinous neoplasm (IPMN) and 30 healthy controls and reported that plasma miR--‐21 levels were associated with advanced cancer stage, lymph node and liver metastasis, and shorter survival [102].
Surgical removal of pancreatic cancer lowered circulating miR-221 in 8 patients [103] and miR-18a dropped significantly after surgical removal of pancreatic cancer in 9 patients [104]. Strikingly, at the time of tumor recurrence in one patient, the levels of circulating miR-18a had increased again despite the lack of any elevation of the conventional serum tumor marker CA-19-9. Additionally, circulating miR-483-3p levels decreased after surgery in 2 pancreatic cancer patients [102] and Li et al. [105] report that higher miR-1290 levels predicted poorer outcome among patients undergoing pancreaticoduodenectomy.
Drug therapy impacts many organ systems. MiR concentrations in the circulation could serve as easily accessible markers of treatment efficacy and even indicate pathways altered by a given treatment. Wang et al. showed that drug induced liver injury can be indicated by a dramatic increase in plasma miR-122 and decrease in miR-170. The circulating miRs showed to be more sensitive markers than alanine aminotransferase for liver injury [106]. We studied circulating miRs before and after Gemcitabine treatment in healthy and pancreatic cancer-bearing mice [107]. MiR-10, miR-21 and miR-155 serum levels decreased significantly after Gemcitabine treatment in both healthy mice and in KrasG12D driven pancreatic cancer mice. Interestingly, serum miR-155 concentrations in mice with pancreatic cancer were significantly lower than in control mice upon Gemcitabine treatment suggesting miR-155 as a potential indicator of tumor specific effects of the treatment.
In a report by Shivapurkar et al. we showed that circulating miR-296 is lost during tumor progression and correlates with metastatic disease in colorectal cancer [108]. Patients with metastatic colon cancer were treated with a multi-targeted receptor tyrosine kinase inhibitor, Sunitinib, and the anti-metabolite Capecitabine. Circulating miRs were analyzed from 7 pre- and matching post-treatment serum samples. 3 patients had decreased miR-296 at 4 weeks post treatment. 4 patients had an increase in the level of miR-296 during that time period. Compared to patients with longer survival and better clinical outcome, patients with shorter survival and poor clinical outcome exhibited a decrease in the level of miR-296 at 4 weeks compared to baseline. The loss of miR-296 may be one of the mechanisms for primary resistance of colorectal cancer to chemotherapy and this could be translated to studies in pancreatic cancer patients using similar drug regimen.
"Liquid biopsies", a recently adopted term for blood based molecular analyses in cancer patient diagnostics can provide innovative monitoring of the evolution of the disease and the response to treatment. Both circulating microRNA and cancer cell derived mutant DNA have been tested as potential surrogates or complements for direct tissue biopsies. A major strength of liquid biopsies is the possibility to compare serial samples from the same patient and thus generate a molecular readout of disease progression and therapy response or resistance in real time. Also the blood draw is minimally invasive and provides bio specimen of comparable composition from a homogeneous compartment, i.e. the blood stream.
Mutant cfDNA and circulating miRs provide distinct and complementary information: Alterations in cfDNA mutation patterns originate from treatment resistant cancer cell subpopulations and their emergence could prompt changes in treatment. On the other hand, altered patterns of circulating miR expression indicate a change in the steady state of the whole organism that carries the disease in response to treatment as well as disease response or progression. Thus, we propose to use a combination analysis of circulating mutant DNA and microRNAs to monitor treatment responses in patients with pancreatic cancer (Figure 1). However, sensitive detection methods and extensive analyses are needed to uncover indicators of diseased organs in a background of circulating nucleic acids from healthy organs. Figure 1 illustrates this challenge and the opportunity to provide deeper insights into the molecular alterations driving pancreatic cancers.
We would like to thank Dr. Narayan Shivapurkar who shared his experience in isolating and measuring circulating miRs, Dr. Ivana Peran for her thoughts on the use of circulating miRs as drug response markers, Jeroen Versteeg, for discussions on circulating miRs in patients with pancreatic cancer and Dr. Niels Kok for discussions on clinical utility of circulating nucleic acids in cancer patients. Work was supported in part by NIH/NCI grants R01 CA71508 (AW), R33 CA177466 (EEV, AW) and P30 CA51008 (AW) as well as the Ruesch Center at Georgetown University (AW).