Journal of Drug Metabolism & Toxicology
Open Access
ISSN: 2157-7609
ISSN: 2157-7609
Research Article - (2016) Volume 0, Issue 0
Keywords: Batracylin; N-Acetylbatracylin; Metabolism; Microsomes; Hepatocytes
BAT: Batracylin; BNPP: Bis(4-Nitrophenyl)-Phosphate; DEX: Dexamethasone; DLM: Dog Liver Microsomes; GSH: Glutathione; HLM: Human Liver Microsomes; HPLC: High-Pressure Liquid Chromatography; LC-MS: Liquid Chromatography-Mass Spectrometry; 3MC: 3-Methylcholanthrene; NAB: N-Acetylbatracylin; NADPH: Nicotinamide-Adenine Dinucleotide Phosphase (reduced); NAT: N-Acetyltransferase; PB: Phenobarbital; RLM: Rat Liver Microsomes
Batracylin (BAT) is a heterocyclic arylamine that was shown to decrease tumor growth in preclinical tumor models [1-3]. Mechanistic studies demonstrated that BAT is an ATP-insensitive, dual inhibitor of topoisomerases I and II [4]. In the course of preclinical development, it became apparent that there is significant interspecies variation in both the toxicity and pharmacokinetics of BAT. Rats were considerably more sensitive to its toxic effects than dogs, with an oral maximum tolerated dose (mg/m2) ratio of 60X, whereas mice demonstrated an intermediate sensitivity [5,6]. Dose-limiting toxicities in rats were to kidney and bone marrow, with lesser toxicities to liver, gastrointestinal tract, and testis. BAT is extensively metabolized in rats by NAcetyltransferases (NAT) to N-Acetylbatracylin (NAB), whereas the NAB exposure from administered BAT is 10-fold less in the mouse [6]. NAB was not observed at all in dog plasma following oral administration of BAT [7], which is consistent with the reported lack of NAT expression in this species [8]. These observations led to the hypothesis that BAT toxicity is dependent on the formation of NAB, a known toxic molecule [9], in sensitive species. Based on this hypothesis, a Phase I clinical trial of BAT was initiated at the National Cancer Institute in patients with advanced cancer and a pharmacogenetically documented NAT2 slow-acetylator genotype (NCT00450502) [10]. However, unexpected dose-limiting toxicity of cystitis/hemorrhagic cystitis was observed and clinical development of BAT was discontinued in the absence of strong efficacy signals [10].
To further understand the metabolism of BAT and its relationship to the observed species differences in toxicity, we conducted a series of in vitro studies to characterize species-specific metabolism in rat, dog, and human liver preparations (microsomes and hepatocytes), and with recombinant cytochrome P450 isoforms. Oxidative metabolites and thiol conjugates of BAT and NAB were formed in multiple incubation systems. As these observations were consistent with metabolic activation of BAT to potentially reactive intermediates, covalent binding of [14C]BAT and [14C]NAB to protein and DNA were also assessed. NADPH-dependent BAT and NAB covalent binding were detected in multiple microsomal preparations, suggesting CYPcatalyzed oxidation of BAT yields reactive metabolites that bind protein and DNA and may contribute to drug-related toxicities.
Compounds and chemicals
BAT (NSC 320846), NAB (NSC 611001), N-propyl BAT, 14C-BAT and differentially deuterated (d3- and d4-) BAT were provided by the Developmental Therapeutics Program, DCTD, NCI. 14C-NAB, d3- NAB, and d4-NAB were prepared in situ by incubating BAT with NAT2 and acetyl-CoA. β-Nicotinamide adenine dinucleotide phosphate, reduced form 95% (NADPH), acetyl coenzyme A sodium salt (acetyl-CoA), DL-dithiothreitol, Bis(p-nitrophenyl) phosphate sodium salt (BNPP), Tris-HCl, L-glutathione reduced form (GSH), 0.5% triton X-100 and ammonium acetate were purchased from Sigma Aldrich (St. Louis, MO). Acetonitrile was purchased from Fisher Scientific (Fairlawn, NJ). HPLC grade water, methanol, and sodium hydroxide (NaOH) were purchased from EMD (Billerica, MA). Bovine serum albumin standard was purchased from Thermo Scientific (Waltham, MA). Ultrapure salmon sperm DNA was purchased from Invitrogen (Grand Island, NY). Ultima gold scintillation fluid was purchased from Perkin Elmer (Waltham, MA).
Microsomes
Pooled human liver microsomes (20 mg protein/mL, 250 mM sucrose; HLM) and human NAT2 cytosol (2.5 mg protein/mL) were obtained from BD Biosciences (Woburn, MA). Rat Liver Microsomes (RLM) were obtained from Celsis (Baltimore, MD). The following preparations were used: Sprague-Dawley male RLM, dexamethasoneinduced Sprague-Dawley male RLM, 3-methylcholanthrene-induced Sprague-Dawley male RLM, phenobarbital-induced Sprague-Dawley male RLM, and Fischer 344 male RLM. Male beagle dog liver microsomes (24.8 mg/protein/mL, 250 mM sucrose; DLM) were obtained from Celsis (Baltimore, MD).
cDNA-expressed P450 enzymes
Microsomal suspensions were obtained from BD Biosciences (Woburn, MA). Microsomes for 11 human P450s (CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C9A, CYP2C19, CYP2D6, CYP2E1 and CYP3A4) and 2 rat P450s (CYP2A1 and CYP2E1) were prepared in AHH-1 TK+/- B-lymphoblastoid cell lines. Microsomes from non-transfected cells and cells that contained the expression vector alone were used as controls. Microsomes for 2 human P450s (CYP1A1 and CYP1B1) and 9 rat P450s (CYP1A1, CYP1A2, CYP2A2, CYP2B1, CYP2C6, CYP2C13, CYP2D1, CYP3A1, or CYP3A2) were prepared in baculovirus-infected insect cells (BTITN- 5B1-4). Microsomes from the insect cells infected with wild type baculovirus were used as controls for those P450s.
Microsomal incubation conditions
Incubations of microsomal suspensions were performed in 2 mL slick microcentrifuge tubes maintained at 37°C in a thermomixer. Each 400 μL incubation mixture contained RLM, HLM, or DLM (0.4 mg protein), NADPH (1.0 mM), magnesium chloride (5 mM) and potassium phosphate (100 mM) buffer adjusted to pH 7.4. Control incubation mixtures contained active microsomes without added NADPH. BAT or NAB (final concentration 10 μM) was added to the incubation mixture after 2-minute pre-incubation. Aliquots (50 μL) were removed at indicated time points and reactions were terminated by addition to 50 μL ice-cold methanol containing internal standard (4.0 μM). The aqueous methanol supernatants obtained following centrifugation (14,000 rpm for 5 minutes) were placed in autosampler vials and analyzed by LC-MS. For GSH metabolism, GSH (10 mM) was incubated with microsomal suspensions as above. Recombinant CYP suspensions were prepared in the same manner as microsomal suspensions, with a P450 concentration of 50 pmol/mL.
In situ acetylation of BAT
To convert BAT to NAB, incubations with NAT2 cytosol and acetyl CoA were performed in 2 mL slick microcentrifuge tubes maintained at 37°C in a thermomixer. Each 500 μL incubation mixture contained Tris assay medium (Tris pH 7.4 (20 mM), DL-dithiothreitol (0.1 mM), acetyl CoA (2 mM)), and NAT2 cytosol (25 μg). BAT (final concentration of 10 μM) was added to the incubation mixture after a 2-minute pre-incubation, and incubated for an additional 30 minutes to form NAB. For P450 metabolism studies, microsomes (0.5 mg protein), BNPP (0.1 mM) and NADPH (1 mM) were incubated for an additional 120 minutes. BNPP was added to inhibit NAB deacetylation to BAT. Cofactors acetyl-CoA and NADPH were omitted in control incubations for acetylation and P450 metabolism. All reactions were terminated by the addition to 500 μL ice-cold methanol. The aqueous methanol supernatants obtained following centrifugation (14,000 rpm for 5 minutes) were placed in autosampler vials and analyzed by LCMS.
Hepatocytes
Pre-plated fresh Sprague-Dawley rat and human hepatocytes with Geltrex overlay were obtained from CellzDirect (Durham, NC). 24- well plates were received on ice 48 hours post isolation in cold preservation medium. Williams E medium and cell maintenance supplement pack were obtained from the vendor, prepared as per vendor instructions, sterile filtered, and stored at 4°C protected from light. Medium was changed upon receipt to complete culture medium and allowed to incubate overnight. Rat hepatocyte viability was 93%, while human hepatocyte viability was 85-94%. Beagle dog hepatocytes were purchased from Celsis in vitro Technologies (Baltimore, MD). Fresh, male, beagle dog plated hepatocytes were isolated by the vendor and suspended in in vitro Gro Incubation medium with phenol red (Celsis). Cells were plated at 350,000 cells/well in collagen coated, 24- well plates and given a matrigel overlay. Plates were shipped (on ice) from the vendor, received the next day and incubated (37°C/5% CO2) overnight.
Incubation conditions for fresh human and rat hepatocytes
BAT stocks (14C-labeled and non-radiolabeled) were diluted with DMSO, then added to complete medium, to yield BAT concentrations of 0, 0.5, 2.5, or 10 μM with 0.25% DMSO. After aliquots (200 μL) of drug containing medium were removed to determine initial incubation concentrations, medium was aspirated from the wells containing the rat hepatocytes and replaced with freshly prepared drug containing medium (700 μL). All drug concentrations were run in triplicate. Aliquots (200 μL) of medium were removed from the wells after 1, 3, and 6 hr, snap frozen on dry ice and stored at -70°C until worked up for analysis. After drug exposure, remaining medium was aspirated, cells were washed once with phosphate buffered saline (2 mL) then lysed with ice-cold 0.5% Triton X-100 (200 μL). Protein was collected and quantified by the DC assay (Bio-Rad). Prior to analysis, aliquots were thawed, diluted 1:1 with methanol, placed in autosampler vials with glass inserts and analyzed by HPLC with radiochemical detection (14C-BAT incubations) or LC/MS/MS (non-radiolabeled BAT incubations).
Incubation conditions for fresh dog hepatocytes
After receipt and overnight incubation, medium was replaced with fresh medium containing vehicle (0.1% DMSO), vehicle + BAT (0.5, 5, and 25 μM), or vehicle + NAB (0.5, 2.5, or 10 μM). Each treatment was run in triplicate wells. Plates were returned to the incubator and removed at 1, 3, or 6 hr post drug exposure. At harvest, plates were removed and 200 μL/well of treatment medium was removed and all three wells’ medium was pooled into a cryovial (600 μL total). Cryovials were snap frozen in a mixture of methanol and dry ice, and vials were stored at -80°C until analysis.
Protein and DNA binding
Metabolic activation and subsequent covalent binding was measured as the amount of radioactivity retained by microsomal protein or exogenously added salmon sperm DNA after extensive washing with organic solvents. To measure BAT covalent binding, 14CBAT (10 μM, 0.1μCi/500μL) was added to microsomal incubations. To measure NAB covalent binding, 14C-BAT (10 μM, 0.1μCi/500μL) was converted to 14C-NAB using NAT2 cytosol procedure described above. Following incubations of radiolabeled BAT and NAB with microsomal preparations, reactions were quenched with acetone. Precipitated protein was washed with 80% methanol until no further decrease in radioactivity was detected in supernatant washes. Air-dried pellets were mixed with 1N NaOH and heated at 60°C for 1 hr to hydrolyze proteins. Protein concentrations were determined using the DC protein assay using bovine serum albumin for the standard curve. Samples were prepared for scintillation analysis by neutralizing with 4N HCl, mixing with ultima gold scintillation fluid, and letting quench in the dark for 60 min. Radioactivity was measured by scintillation analysis, corrected for background quenching (external standardization), and binding was expressed as nmol drug equivalents per milligram protein. Microsomes were boiled for 10 minutes at 100°C and used as controls for protein binding. To measure DNA binding, salmon sperm DNA (100 μg) was added to microsomal suspensions. After a 120-min incubation, reactions were quenched with methanol and the resulting precipitate was washed with phosphate buffered saline. DNA was isolated using a DNeasy Blood and Tissue kit (Qiagen). DNA concentrations were measured using the A260/A280 UV absorbance method. Samples were prepared for scintillation analysis by mixing with ultima gold scintillation fluid and letting quench in the dark for 60 min. Radioactivity was measured by scintillation analysis and binding was expressed as nmol drug equivalents per milligram DNA.
Instrumentation
The HPLC system consisted of a Shimadzu high pressure liquid chromatograph (HPLC) system (Wood Dale, IL, USA), containing two LC-10AD pumps with a semi micro mixer, a SIL-10ADvp autosampler, SPD-M20A diode array detector in tandem with RF535 fluorescence detector or Packard 500TR series flow scintillation analyzer (Waltham, MA). The LC-MS system consisted of the Shimadzu HPLC coupled to a triple quadrupole Quattro Micro mass spectrometer (Waters, Milford, MA, USA) equipped with an electrospray ionization (ESI) source. Data was acquired and analyzed by Shimadzu Class-VP version 7.2 SP2 (HPLC), Perkin Elmer PROFSA version 3.4.3.36b (HPLCscintillation analyzer) or Waters MassLynx v4.1 (LC-MS) software.
Chromatographic conditions
The liquid chromatographic separation of BAT, NAB, and its metabolites were accomplished using a Brownlee newGuard RP-18 precolumn (Chrom Tech, Apple Valley, MN) attached to a Vydac Genesis C18 analytical column (Chrom Tech, Apple Valley, MN), eluted with a isocratic mobile phase composed of 23:77 acetonitrile:NH4OAc, pH 3.5 with a constant flow rate of 0.5 mL/min and a total run time of 24 min. Autosampler temperature was 25°C and sample injection volume was 30 μl.
HPLC detection of BAT, NAB, and its metabolites were accomplished using the diode array detector wavelength range 240-450 nm and specific wavelength 413 for data collection and the fluorescence detector with set wavelengths 390 for excitation and 565 for emissions. For radiochemical detection scintillation flow rate was set to 2.0 mL/min and channel was set to detect 14C compounds.
LC-MS detection of BAT, NAB, and its metabolites were accomplished using the mass spectrometer in positive ESI mode using capillary voltage 3.40 kV, source temp 120°C, desolvation temp 350°C, cone gas flow 25 L/hr, desolvation gas flow 650 L/hr, using Multiple Reaction Monitoring (MRM), full scan, and product ion scan modes with a dwell time of 0.25 sec. The cone voltages and collision energies varied between 30-85 V and 32-40 eV, respectively. The MRM precursor and product ions were monitored at m/z 250 > 130 for BAT, 292 > 130 for NAB, 253 > 130 for d3-BAT (internal standard), and 254 > 134 for d4-BAT. Full scan was monitored at m/z range 30-750.
Stock solution preparation
The primary stock solutions of BAT (2.0 mM), NAB (1.72 mM), d3- BAT (1 mM), d4-BAT (1 mM), and 14C BAT (4 mM, 0.08μCi/μL) were dissolved in DMSO and stored at -20°C. Working standards were prepared daily by dilution of the stock solution with 1:1 methanol:H2O.
Metabolism in human liver microsomes
HPLC chromatograms illustrating the NADPH-dependent loss of BAT and NAB in HLM are shown in Figure 1.

Figure 1: UV chromatograms showing metabolites formed after 120 minute incubation of human liver microsomes in the presence of A) 10 μM BAT without NADPH or B) 10 μM BAT with NADPH C) 10 μM NAB without NADPH or D) 10 μM N-acetyl BAT with NADPH. E) 10 μM NAB + 0.1 mM BNPP without NADPH. F) 10 μM NAB + 0.1 mM. BNPP with NADPH. BNPP inhibits conversion of NAB to BAT in active HLM.
BAT was reduced by 30% (Figure 1A and 1B) and NAB was reduced by 36% (Figure 1C and 1D) after 120-minute incubation with NADPH-fortified pooled HLM. NADPH-dependent loss of BAT was accompanied by formation of a product identified by LC/MS analysis as hydroxylated BAT based on the presence of a molecular ion with m/z 266 (M+16) in ion chromatograms (Figure 2A).

Figure 2: Metabolites formed after 120 minute incubation of BAT (10 μM) with HLM. A) LC-MS chromatogram of HLM incubated with BAT. Top: MS scan m/z 266 -NADPH. Bottom: MS scan m/z 266 + NADPH; B) LC-MS chromatogram of HLM +NADPH incubated with d3-BAT for 120 min MS scan m/z 268 and C) LC-MS chromatogram of HLM +NADPH incubated with d4-BAT for 120 min MS scan m/z 270.
Microsomal incubations with differentially-labeled d3- and d4- BAT indicated that this metabolite was produced by C-hydroxylation in the aromatic amine ring of BAT (metabolite B2 in Table 1) based on detection of a product with m/z 268 (Figure 2B) in incubations with d3-BAT and a product with m/z 270 (Figure 2C) in incubations with d4-BAT.
| Metabolite | Retention time (min) | Molecular Ion | Structure | Samples metabolite is found in |
|---|---|---|---|---|
| B1 | 6.4-6.8 min | d0- m/z 266 d3- m/z 269 d4- mz/ 269 | 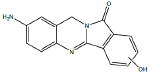 |
3MC RLM SD RLM Pb RLM Fisher RLM Dex RLM H. CYP1A1 |
| B2 | 7.5-9.0 min | d0- m/z 266 d3- m/z 268 d4- m/z 270 | 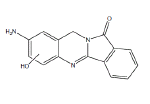 |
SD RLM Pb RLM Fisher RLM Dex RLM 3MC RLM HLM H. CYP1A1 H. CYP1A2 H. CYP1B1 R. CYP1A1 R. CYP1A2 Dog- hepatocytes 3MC RLM+GSH HLM+GSH |
| B3 | 9.1 min | d0- m/z 266 d3- m/z 269 d4- mz/ 270 | 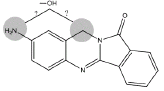 |
3MC RLM |
| B4 | 10.5-11.5 min | d0- m/z 266 | Unknown | H. CYP1A1 H. CYP1B1 R. CYP1A1 R. CYP1A2 3MC Rat+GSH |
| B5 | 3.0-3.7 min | d0- m/z 282 d3- m/z 284 d4- mz/ 286 |
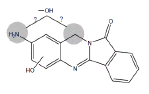 |
HLM3MC RLM R. CYP1A1 H. CYP1A1 H. CYP 1B1 3MC RLM+GSH HLM+GSH |
| B6 | 4.8-6.3 min | d0- m/z 282 d3- m/z 284 d4- mz/ 286 |
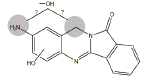 |
3MC RLM H. CYP1A1 |
| N1 | 11.2-12.2 min | d0- m/z308 d3- m/z311 d4- m/z312 | 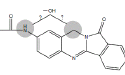 |
3MC RLM H. CYP1A1 H. CYP1A2 R. CYP1A1 R. hepatocytes |
| N2 | 13.0-14.0 min | d0- m/z308 d3- m/z310 d4- m/z312 | 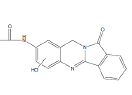 |
3MC RLM H. CYP1A1 R. CYP1A1 R. CYP1A2 R. hepatocytes D. hepatocytes 3MC RLM+GSH HLM+GSH |
| N3 | 16.5-18 min | d0- m/z308 d3- m/z311 d4- m/z312 | 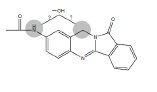 |
3MC RLM H. CYP1A1 R. CYP1A1 3MC RLM+GSH |
| N4 | 3.5-4.4 min | d0- m/z324 d3- m/z326 d4- m/z328 | 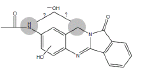 |
3MC RLM H. CYP1A1 H. hepatocytes |
Table 1: Proposed Structures for BAT and NAB oxidative (Phase I) metabolites.
NADPH-dependent loss of NAB was accompanied by formation of hydroxylated BAT, BAT, and trace amounts of other products (Figure 1D). The loss of NAB (24%) observed in control incubations that did not contain NADPH (Figure 1C) was attributed to metabolism by microsomal carboxylesterase because the carboxylesterase inhibitor BNPP substantially reduced formation of BAT in microsomal incubations with NAB (Figure 1C vs. 1E), without altering BAT metabolism in parallel incubations (data not shown).
Metabolism in human CYP-expressing microsomes
BAT and NAB were incubated with a panel of microsomes expressing individual human CYP enzymes. BAT was primarily metabolized by human CYP1A1, CYP1A2, and CYP1B1, as shown in Figure 3A, 3B and 3C, respectively. The percentages metabolized in 120 min incubations are shown in Table 2.

Figure 3: UV chromatograms showing metabolites formed after 120 minute incubation with 10 μM BAT (A-C) NAB (D-F) in the presence of NADPH-fortified human cDNA expressing microsomes: CYP1A1 (A and D), CYP1A2 (B and E), CYP1B1 (C and F).
| Enzyme | BAT | NAB |
|---|---|---|
| % Metabolized in 120 min | ||
| Human CYP-expressing microsomes | ||
| 1A1 | 18 | 62 |
| 1A2 | 8.4 | 8 |
| 1B1 | 32 | 17 |
| 2E1 | <3 | 7 |
| Others | <3 | <1 |
| Rat CYP-expressing microsomes | ||
| 1A1 | 34 | 33 |
| 1A2 | 31 | 27 |
| Others | <3 | <1 |
| Induced rat microsomes* | ||
| Untreated | 22 | 12 |
| DEX (3A) | 23 | 12 |
| 3MC (1A) | 70 | 79 |
| PB (3A, 2B) | 59 | 10 |
Table 2: Metabolism of BAT and NAB by Microsomal Preparations. *Liver Microsomes from rats pretreated with dexamethasone (DEX), 3-methlycholanthrene (3-MC) or phenobarbital (PB), showing the primary CYPs induced by these treatments.
The remaining CYPs tested reduced BAT by less than 3%. Mono and di-hydroxy-BAT were detected in each reaction, with the greatest formation observed in CYP1B1 incubations (Figure 3C, Supplementary Figure 1).
NAB was metabolized by microsomes expressing human CYP1A1, CYP1A2, CYP1B1, and CYP2E1 as illustrated in Figures 3D, 3E and 3F, respectively, and the percentages in Table 2. The remaining CYPs metabolized NAB by less than 1%. Multiple oxidative metabolites were detected in incubation aliquots, including a predominant metabolite (N3) formed with CYP1A1 and CYP1B1 that eluted just prior to the NAB peak (Figures 3D and 3F, respectively).
Metabolism in RLM
BAT and NAB were incubated with a series of liver microsomal preparations from Sprague-Dawley male rats, including untreated rats, dexamethasone (DEX)-pretreated rats that highly express CYP3A isoforms, 3-methylcholanthrene (3MC)-pretreated rats that highly express CYP1A isoforms, and phenobarbital (PB)-pretreated rats that highly express CYP3A and CYP2B isoforms.
The BAT concentration was reduced in each of these NADPHfortified microsomal incubations (Figure 4A, 4B, 4C and 4D, respectively) by the percentages shown in Table 2. Mono- and dihydroxy- BAT were detected in each of these reactions with the greatest formation observed in 3MC incubations (Figure 4C, Supplementary Figure 2). NAB was metabolized by NADPH-fortified RLM preparations (Figure 4E, 4F, 4G, and 4H) by the percentages shown in Table 2. Mono- and di- hydroxyl-NAB were detected in each of these reactions with the greatest formation observed in the 3MC incubations based on the UV chromatogram and molecular ions with m/z 308 (monohydroxy-NAB) and m/z 324 (dihydroxy-NAB) (Figure 4G, Supplementary Figure 3).

Figure 4: UV chromatograms showing metabolites formed after 120 minute incubation with 10 μM BAT (A-D) or NAB (E-H) in the presence of NADPH-fortified Sprague-Dawley male RLM: No pretreatment (A and E), dexamethasone pretreatment (B and F), 3-methylcholanthrene pretreatment (C and G), phenobarbital pretreatment (D and H).
Metabolism in rat CYP expressing microsomes
BAT and NAB were incubated with a panel of microsomes expressing individual rat CYP enzymes. BAT was metabolized by rat CYP1A1 and CYP1A2 (Supplementary Figures 4A and 4B, respectively), whereas the remaining CYPs metabolized BAT by less than 3% (Table 2). Monohydroxy-BAT metabolites were detected in both reactions, while dihydroxy-BAT was only detected in the rat CYP1A1 reaction based on the UV (Supplementary Figure 4A and 4B) and ion (data not shown) chromatograms. NAB was metabolized only by microsomes expressing rat CYP1A1 and CYP1A2 (Table 2 and Supplementary Figures 4C and 4D). Mono- and di- hydroxyl-NAB were detected in the rat CYP1A1 reaction, while monohydroxy-NAB was detected in the rat CYP1A2 reaction based on the UV (Supplementary Figure 4C and 4D and ion (data not shown) chromatograms.
Metabolism in DLM
Metabolism of BAT and NAB was investigated with DLM to compare metabolite formation with that observed for RLM and HLM. NADPH-dependent loss of BAT was accompanied by formation of a product identified as monohydroxy-BAT based on the presence of a molecular ion with m/z 266 in ion chromatograms and a similar retention time compared with that observed in HLM and RLM (Supplementary Figure 5). NADPH-dependent loss of NAB in DLM incubations containing BNPP was accompanied by formation of products identified as BAT and dihydroxy-NAB (Supplementary Figure 6). A trace amount of BAT was also detected following incubation in the absence of NADPH.
Glutathione-S-Transferase catalyzed metabolism of BAT and NAB
BAT and NAB were incubated with pooled HLM or 3-MC induced RLM fortified with NADPH and GSH. In addition to the hydroxylated metabolites observed in earlier in vitro metabolism studies, we also detected BAT (m/z 555), hydroxy BAT (m/z 571) and NAB (m/z 597) thiol conjugates (Table 3).
| Metabolite1 | (m/z) | Retention Time (HPLC) (min) | Metabolite Medium |
|---|---|---|---|
| BAT+NAC | 411 | 3.0-3.9 | human and rat hepatocytes, 3MC RLM+GSH, HLM+GSH |
| BAT+Gluc | 426 | 3.5-4.3 | human, rat and dog hepatocytes |
| BAT+OH+NAC | 427 | 3.6-4.1 | rat and dog hepatocytes |
| BAT+GSH | 555 | 3.0-3.8 | human and dog hepatocytes |
| BAT+GSH | 555 | 4.5-5.3 | human hepatocyte, HLM+GSH, 3MC RLM+GSH |
| BAT+GSH | 555 | 18.4 | rat hepatocytes |
| BAT+OH+GSH | 571 | 3.1-4.0 | HLM+GSH, 3MC RLM+GSH |
| NAB+GSH | 597 | 3.4-3.8 | Dog hepatocytes, 3MC RLM+GSH, HLM+GSH, rat kidney |
Table 3: BAT and NAB Phase II metabolites. 1Abbreviations: BAT: Batracylin; NAC: N-Acetyl-Cysteine, Gluc, Glucuronide; GSH: Glutathione.
Structural identification of metabolites in microsomal incubations
Efforts to assign structures to oxidative metabolites of BAT focused on major products formed by incubation of BAT, d3-BAT, and d4-BAT with human CYP1B1 (Figure 3C) and 3MC RLM (Figure 4C) with comparisons of precursor and product ions of non-deuterated and deuterated metabolites. Of note, retention times on LC/MS/MS chromatograms are approximately 2 minutes later than the retention times on the HPLC/UV chromatograms.
Metabolite B1 (Table 1) appears to result from aromatic carbon hydroxylation in the indole ring of BAT, based on comparison of the precursor ion m/z 266 and product ion m/z 146 with monohydroxylated d3-BAT which had a precursor ion of m/z 269 and product ion m/z 146, and monohydroxylated d4-BAT which had a precursor ion of m/z 269 and product ion m/z 149 indicating hydroxylation of an aromatic carbon in the indole ring of BAT (Figure 5). The location of the hydroxylation in the indole ring cannot be determined from these data.

Figure 5: Metabolite B1- BAT (m/z 266), d3-BAT (m/z 269) and d4-BAT (m/z 269) incubated with 3MC-induced RLM.
Metabolite B2 (Table 1) is the major oxidative BAT metabolite formed in microsomal incubations and appears to result from aromatic ring hydroxylation of BAT by comparison of the precursor ion m/z 266 and product ions m/z 130 and 249 for d0-BAT metabolite with the precursor ion of m/z 268 and product ions m/z 130 and 251 for the d3- BAT metabolite, and the precursor ion of m/z 270 and product ions m/z 134 and 253 for the d4-BAT metabolite. These data are consistent with hydroxylation of an aromatic carbon in the aniline ring of BAT (Figure 6). The location of the hydroxylation in the aromatic ring cannot be determined from this data.

Figure 6: Metabolite B2- BAT (m/z 266), d3-BAT (m/z 269) and d4-BAT (m/z 270) incubated with 3MC-induced RLM.
Metabolite B3 (Table 1) appears to be the result from the hydroxylation of either the amine nitrogen or the methylene carbon (Figure 7). The hydroxylation site for B4 could not be determined from these data.

Figure 7: Metabolite B3- BAT (m/z 266), d3-BAT (m/z 269) and d4-BAT (m/z 270) incubated with 3MC induced RLM.
Metabolites B5 and B6 (Table 1) have been identified as dihydroxy BAT based on the presence of m/z 282 in the ion chromatograms indicating addition of two oxygen atoms to BAT. Microsomal incubations with d3- and d4-BAT indicated that these metabolites result from one hydroxylation on the aromatic amine ring moiety of BAT, but it has not been possible to determine whether the second hydroxylation occurs at the amine nitrogen or the methylene carbon (Table 1).
Efforts to assign structures to oxidative metabolites of NAB focused on products formed by incubation with human CYP1B1 (Figure 3D) and 3MC RLM (Figure 4G), where four peaks were detected on HPLC/UV chromatograms, and comparison of precursor and product ions in non-deuterated and deuterated metabolites from NADPH containing 3MC RLM, rat CYP1A1, and human CYP1A1. Metabolites N1, N2 and N3 (Table 1) were identified as mono-hydroxylated metabolites of NAB based on addition of oxygen (+16) to yield molecules with m/z 308. Using d3- and d4-NAB prepared in situ , we confirmed that N2 was formed by hydroxylation of NAB in the aromatic amine ring (Figure 8). The location of hydroxylations for N1 and N3, which appear to be formed by hydroxylation of the aromatic amine or benzylic methylene groups, could not be confirmed (data not shown). The N4 (Table 1) metabolite was identified as dihydroxyl NAB based on the presence of m/z 324 precursor ion. The presence of an m/z 130 fragment in mass scans for this peak, consistent with the oxoindole moiety, suggests that both hydroxylations are in the aromatic amine ring of NAB. We were not able to determine whether one of the hydroxylations is on the aromatic amine nitrogen and our efforts to identify the NAB hydroxylation sites, using d3- and d4-NAB prepared in situ by acetylation of d3-BAT, and d4-BAT as described above, were unsuccessful.

Figure 8: Metabolite N2- NAB (m/z 308), d3-NAB (m/z 310) and d4-NAB (m/z 312) incubated with 3MC-induced RLM and rat CYP1A1.
Hepatocyte incubations
Human hepatocytes: Incubations of 0.5, 2.5 and 10 μM 14C-BAT with human hepatocytes from three separate donor livers resulted in formation of two main metabolite peaks that accounted for all of the radioactivity in radiochromatograms (Supplementary Figure 7A) and the major peaks detected in UV absorbance chromatograms (Supplementary Figure 7B). As illustrated in an ion chromatogram for an aliquot from a parallel incubation with unlabeled BAT (Supplementary Figure 7C), the peaks were identified as BAT Nglucuronide, based on detection of a product ion with m/z 426 (+176 resulting from addition of glucuronic acid to BAT) and NAB based on co-elution with the authentic standard and LC-MS analysis. Trace amounts of hydroxy-BAT (m/z 266) and N-acetylcysteine BAT (m/z 411) were also detected in the human hepatocyte samples.
Rat hepatocytes: Incubations of 0.5, 2.5 and 10 μM 14C-BAT with fresh rat hepatocytes resulted in formation of two main metabolite peaks that accounted for all of the radioactivity in radiochromatograms (Supplementary Figure 8A) and the major peaks detected in UV absorbance chromatograms (Supplementary Figure 8B). As illustrated in an ion chromatogram for an aliquot from a parallel incubation with unlabeled BAT (Supplementary Figure 8C), the peaks were identified as BAT N-glucuronide based on detection of a product ion with m/z 426 (+176 resulting from addition of glucuronic acid to BAT) and NAB based on co-elution with LC-MS analysis of authentic BAT and NAB.
Following incubation of 5 and 25 μM unlabeled BAT with rat hepatocytes, small amounts of several additional metabolites were detected, including hydroxy BAT (m/z 266) and hydroxy NAB (m/z 308), as well as the N-acetylcysteine conjugates of BAT (m/z 411) and hydroxy BAT (m/z 427) (not shown).
Dog hepatocytes: Samples of the incubation medium and cells from studies with dog hepatocytes from were analyzed for BAT, NAB and their metabolites. BAT glucuronide (m/z 426), was detected in incubations with 25 μM BAT (Supplementary Figure 9). There were no NAB metabolites when NAB was incubated with dog hepatocytes (not shown).
Metabolite profile summary
The metabolites found under the various conditions reported herein are summarized in Tables 1 and 3 for Phase I and Phase II products, respectively.
Metabolic activation of BAT and NAB and binding to protein and DNA
Formation of reactive intermediates, as measured by covalent binding to protein and DNA, was assessed. Human CYP1A1 and CYP1B1 are the major CYP isoforms that metabolize BAT and NAB. These CYP isoforms, as well as HLM, 3MC-RLM, and DLM, were selected for metabolic activation studies. Binding study results are shown in Table 4.
| Protein Binding (pmol/mg) | DNA Binding (pmol/mg) | |||
|---|---|---|---|---|
| BAT | NAB | BAT | NAB | |
| 3MC-RLM | 51 ± 10 | 41 ± 10 | 82 ± 36 | 0 |
| HLM | 22 ± 16 | 43 ±14 | 79 ± 37 | 2 ± 3 |
| DLM | 228 ± 143 | 319 ± 23 | 45 | 16 |
| H. CYP1A1 | 73 ± 27 | 10 ± 12 | 65 ± 25 | 48 ± 22 |
| H. CYP1B1 | 462 ± 151 | 0 | 65 ± 10 | 25 ± 2 |
| 3MC-RLM boiled | 0 | 0 | ND | ND |
| HLM boiled | 4 ± 9 | 0 | ND | ND |
| DLM boiled | 11 ± 11 | 0 | ND | ND |
Table 4: BAT and NAB covalent binding to protein and DNA.
BAT and NAB protein binding was detected at very low levels if at all in incubations with boiled microsomal preparations. The highest protein binding values for 14C-BAT were detected in incubations with human CYP1B1. The highest DNA binding values for 14C-BAT were detected in incubations with 3MC-RLM. The highest protein binding values for 14C-NAB were detected in incubations with DLM, and for DNA binding, in incubations with human CYP1A1.
Toxicological and pharmacokinetic observations during the preclinical development of BAT strongly suggested that the species differences in toxicity, as evidenced by a 60-fold range in oral maximum tolerated dose, are related to the extent of formation of NAB, the N-acetylated metabolite of BAT [5-8]. Based on this hypothesis, a phase I trial of BAT in patients with a slow NAT2 acetylator genotype was conducted [10]. The dose-limiting toxicity was hemorrhagic cystitis; in the absence of objective responses, further development of BAT was halted. Pharmacokinetic analysis showed that plasma exposures to BAT and NAB were similar in these patients [10]. Moreover, human sensitivity to BAT was intermediate along the spectrum from rat (low maximun tolerated dose with high NAB exposure) to dog (high maximum tolerated dose with no NAB exposure), providing further indications that formation of NAB plays a critical role in the toxicity of BAT. However, the mechanism of this differential toxicity (NAB vs. BAT) and potential role of downstream metabolites had not been fully explored.
The studies reported here were undertaken to investigate the metabolic profiles of BAT and NAB using in vitro mouse, rat, dog, and human preparations to identify structures that might be consistent with the differential toxicity (NAB vs. BAT) in these species. While multiple oxidative and conjugated metabolites were identified for both compounds, there was no obvious distinction between BAT and NAB that might explain the differential toxicity across species or dose limiting hemorrhagic cystitis encountered in the clinical trial. The predominant metabolites of BAT in rat, dog, and human liver microsomes are mono-and di-hydroxylated products. Mass spectral analyses indicated that hydroxylations can occur at multiple sites on the molecule (Table 1). The qualitative formation of these mono-and di-hydroxylated species was generally similar across species. In addition, BAT and NAB produced similar metabolite profiles (arylhydroxy and N-hydroxy species) with no clear distinction that might explain the apparent greater toxicity of NAB.
Of note however, some aromatic hydroxylamines are reactive electrophiles that can lead to covalent binding of cellular macromolecules, as was shown for the investigational anticancer compound, aminoflavone [11,12]. To further investigate the formation of reactive intermediates, we measured covalent binding of BAT and NAB to protein and DNA upon incubation with microsomal preparations (Table 3). DNA and protein binding was observed with both compounds, but again there was no clear distinction between compounds or species. The level of DNA binding (μg/mg substrate) was comparable to that reported for aminoflavone [11]. In any case, the data demonstrating both BAT and NAB can be activated to species that bind covalently to DNA provides an avenue for further exploration.
Our studies using microsomes from rats pretreated with one of several CYP-inducing agents demonstrated that the greatest extent of BAT metabolism occurred with 3-methylcholanthrene (3MC)- pretreated animals, in which CYP 1A and 1B isoforms are preferentially overexpressed. Further studies with individual cDNAexpressed human or rat CYP isoforms confirmed the greatest loss of BAT was with 1A and 1B family members. Other CYPs metabolized BAT by less than 3% over the time of incubation. These results support the conclusion that CYPs 1A and 1B are quantitatively the most important oxidative enzymes in the metabolism of BAT in the rat.
These findings differ somewhat from those of Stevens et al. [9], who demonstrated greater cytotoxicity of BAT to hepatocytes from DEXpretreated rats compared with controls. Together with results showing greater toxicity to male- vs. female-derived hepatocytes and a reduction of cytotoxicity in the presence of the CYP inhibitor, troleandromycin, these authors postulated a major role for CYP3A in the metabolism and toxicity of BAT. There are several important methodological differences between this study and ours (induced hepatocytes vs. induced microsomes; loss of substrate endpoint vs. cytotoxicity endpoint; F344 rats vs. Sprague-Dawley rats). While CYP 1A and1B may account for a large fraction of BAT metabolism in rats, it is possible that an unidentified product of CYP3A enzymes contributes disproportionally to toxicity.
A recent investigative toxicology study in rats [13] showed that oral administration of BAT resulted in dose-dependent renal and urothelial (urinary bladder) damage, with frank hematuria in some high-dose animals (it is likely that bladders were not specifically examined in the 1980s toxicology studies). Pharmacokinetic analysis confirmed that NAB was by far the predominant species in plasma, although low levels of BAT and hydroxyl-metabolites of NAB were detected. This study also showed that administration of BAT induced the DNA damage marker γH2AX in kidney and bone marrow tissues. While this effect likely arises from BAT interactions with topoisomerases [4], the consequences of direct covalent binding of “activated” BAT to DNA should be further explored. Results of this toxicology study [13] also argued against a cyclophosphamide-like mechanism of toxicity mediated by production of acrolein, since co-administration of the protective agent, MESNA, did not alleviate BAT-induced renal or bladder toxicity.
Hemorrhagic cystitis has been observed in clinical trials of other topoisomerase inhibitors, such as sodium camptothecin [14], 20-(S)- camptothecin, 9-nitrocamptothecin [15], and etoposide, suggesting it could be a mechanism-based toxicity. Given the prominence of cystitis in the Phase I trial of BAT, topoisomerase inhibition, drug/metabolite distribution to target tissues, and covalent binding of BAT/NAB metabolites may all play a role in this toxicity. An alternative (and speculative) hypothesis for NAB toxicity relates to its poor aqueous solubility (less than BAT itself) and potential precipitation at high concentrations in vivo , with resultant tissue injury. Organ toxicity from the production of a less soluble metabolite is documented for 7-OHmethotrexate [16] and other compounds.
For now, the association of NAB production with renal and bladder toxicity remains provocative but circumstantial. It might seem logical to administer NAB directly to animals and look for its direct and possible differential toxicity across species. Unfortunately, NAB is not absorbed when given orally to rats [6] or dogs and its poor aqueous solubility has impeded studies using intravenous administration. Although the clinical development of BAT was discontinued due to lack of activity and unacceptable toxicity, recent publications [17-19] illustrate that substantial interest remains in designing analogs of BAT with improved pharmaceutical and pharmacological properties. The characterization of BAT metabolites in the present work may provide guidance to this ongoing effort.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. N01-CM-52206. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.