Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 8
LEA proteins are ubiquitous among photosynthetic organisms and have been reported in mono and dicot plants as well as in nematodes, yeast, bacteria and cyanobacteria. EMV2 is a Group 1 LEA protein isolated fromVigna radiata, which is speculated to impart desiccation tolerance in plants. The homology model of this protein was generated by using the LOOPP software based on available structural homologues in protein databases. The final model obtained by molecular mechanics and dynamics method was assessed by PROCHECK that showed that the final refined model is reliable. The model could prove useful in further functional characterization of this protein.
Keywords: LEA proteins, Vigna radiate, Desiccation, Homology modeling, Validation.
SCOP-Structural Classification Of Proteins, PDB-Protein Data Bank, LOOPP -Learning, Observing and Outputting Protein Patterns
Late embryogenesis abundant (LEA) protein genes are highly expressed during late stages of seed development at normal growth condition but many of the LEA class genes are also frequently expressed in vegetative tissues when plants are exposed to environmental stress (Bray et al., 2000). Several groups of LEA protein genes have been demonstrated to confer water-deficit and salt-stress tolerance.
On the basis of sequence similarities, LEA proteins have been classified in to six groups (Dure, 1993; Bray, 1993). Group 2 LEA proteins or dehydrins are by far the most frequently described LEA protein family and have been classified in distinct groups (Close, 1997) that differ in the arrangement and number of conserved motifs: the lysine-rich repeat (KIKEKLPG) or K segment, the stretch of serine or S segment and the V/T DEYGNP motif or Y segment. Some of these structural motifs are predicted to form amphipathic alpha helices, which may be important for their function in protecting plant cells against dehydration. Evidence of functional links between LEA protein accumulation and improved stress tolerance of transgenic yeast and plants support this hypothesis (Imai, 1996; Xu et al., 1996; Sivamani et al., 2000). It was therefore proposed that most LEA and dehydrin proteins exist as largely unfolded structures in their native state, although a few members exist as dimers or tetramers (Ceccardi et al., 1994; Kazuoka and Oeda, 1994). Hydrophilicity is a common characteristic of LEA-type and other osmotic stress-responsive proteins. Our earlier work on the occurrence of LEA proteins in the embryonic axes of Vigna radiata (L.) Wilczek referred as EMV proteins, the first ever report in the Fabaceae family (Manickam and Carlier, 1980). cDNA encoding these proteins were isolated, characterized (Manickam et al., 1996). In silico analysis of the 20-mer motif of this EMV2 categorize this protein to Group1 LEA and hypothesize to function as DNA/RNA binding proteins in stabilizing membranes/macromolecules at the time of dehydration process (Rajesh and Manickam, 2006;Gilles et al., 2007).
In the present study, effort was made to generate the three-dimensional (3D) structure of the EMV2 protein based on the available template structural homologues from Protein Data Bank and SCOP databases and the model validated with standard parameters. This study could prove useful in further functional characterization of this important group of proteins.
Datasets
The peptide sequences of Vigna radiata, EMV2 (NCBI GenBank accession number U31211; UniProt acc. Nos. Q41685 ) and other sequences examined in this study were retrieved from the public databases, http://www.ncbi.nlm.nih.gov and http://www.ebi.ac.uk. Structurally homologous subsets of the experimentally determined 3D structures of the EMV proteins were retrieved from PDB and SCOP databases. The template used for comparative modeling of EMV2 is a DNA Binding Protein from Homo sapiens (1IG6_A.pdb) and a bifunctional inhibitor/ lipid-transfer protein/seed storage 2S albumin (1RZL.pdb) with the sequence similarity of 56.44 and 56.67 % at the loop/coiled coil regions. Similarity at these regions was considered because LEA proteins are generally loosely structured with predominantly random coiled regions. However structure was further refined with sequence of 1RZL.pdb as it showed overall 40 % sequence identity in the sequence length of 89% to a bifunctional inhibitor/lipid transfer protein/ seed storage 2S albumin of rice.
Tertiary structure of the Vigna radiata LEA protein, EMV2 was modeled by submitting the deduced amino acid sequences to the Computational Biology Service Unit, Cornell Theory Center, Cornell University. The Atomic coordinates for the protein models were generated by aligning to the structural homologues in the fold recognition program of LOOPP v3.0 server (Teodorescu et al., 2004).
PROCHECK, a versatile protein structure analysis program (Laskowski et al., 1993) available at the Joint Centre for Structural Genomics, Bioinformatics core, University of California, San Diego was used in validation of protein structure and models by verifying the parameters like Ramachandran plot quality, peptide bond planarity, Bad nonbonded interactions, main chain hydrogen bond energy, Calpha chirality and over-all G factor and the side chain parameters like standard deviations of chi1 gauche minus, trans and plus, pooled standard deviations of chi1 with respect to refined structures( Morris et al., 1992).
Comparative Modeling of EMV2 Protein
Tertiary structure of a protein is build by packing of its secondary structure elements to form discrete domains or autonomous folding units. Comparative modeling to build 3D structure of the EMV2 protein was made based on the experimentally solved structural homologues. The amino acid sequences of EMV2 were submitted to LOOPP server, Cornell Bioinformatics Structural Unit (CBSU) and atomic coordinates for the proteins were generated based on Hidden Markov Model. The hypothetical protein models created were stored as PDB output file. The hypothetical proteins were visualized and computed by Swiss PDB Viewer and Rastop. The 3D structure of the proteins were represented by cartoon display and colored based on the secondary structure (Fig. 1).

Figure 1: Predicted 3-Dimensional Structure of the EMV2 protein. Ribbon view of EMV2 structure for residues 1-99. Numbers represent the order of helices. N and C termini of the protein are labeled.
Validation of Protein Structures of EMV2
The hypothetical protein models generated were analyzed online by submitting to Joint Center for Structural Genomics (JCSG), Bioinformatics core, University of California, San Diego. Accuracy of the protein model generated was judged by validity report generated by PROCHECK. Parameter comparisons of these proteins were made with well-refined structures that have similar resolution.
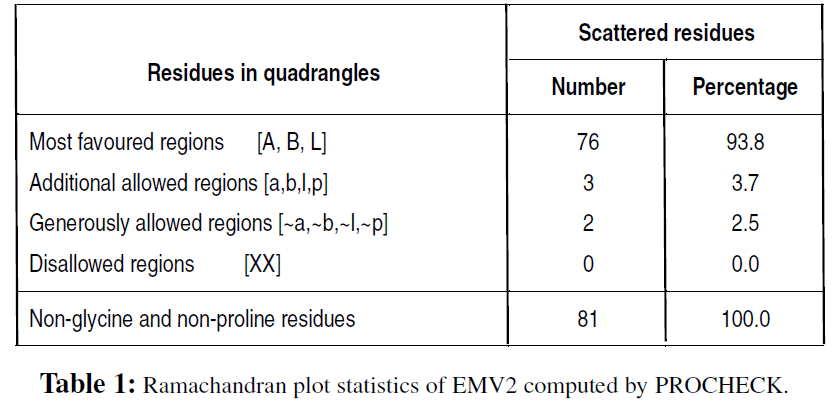
The main chain parameters plotted are Ramachandran plot quality, peptide bond planarity, Bad non-bonded interactions, main chain hydrogen bond energy, C-alpha chirality and over-all G factor. In the Ramamchandran plot analysis, the residues were classified according to its regions in the quadrangle. The Ramachandran map for EMV2 (Fig. 2) and the plot statistics (Table 1) is represented. Non-bonded interactions check revealed the value of 30 bad contacts per 100 residues for EMV2 and the chirality measure of 0.1 for the zeta torsion angle is observed. The over-all G factor computed for the EMV2 is 0.1, which is higher than the ideal value (-0.4).

Figure 2: Ramachandran map of EMV2 protein. The Plot calculation was done with PROCHECK program.
The protein models were analyzed for the side chain parameters like standard deviations of chi1 gauche minus, trans and plus, pooled standard deviations of chi1 with respect to refined structure. The standard deviations of Chi1 gauche minus, trans and plus are in better range and within limits for EMV2 hypothetical protein model.
Computations Performed in the EMV2 Model
Computation of Electrostatic Potential
The electrostatic potential of the residues in the hypothetical protein was computed based on Coulomb’s method. The cloud of charged residues is represented in blue and red colors and the proteins visualized as density map of EMV2 (Fig. 3).

Figure 3: Electron density map of EMV2.
Computation of Force Field Energy
Force field energy was computed for EMV2 protein model. Positive values were observed (64x108 forEMV2). Model refinement was done by energy minimization. Energy minimization was carried out to reduce clashing amino acids, using GROMOS96 force field algorithm. Decrease in the force field energy was observed for both the protein models after successive energy minimization (-1718 for EMV2). The energy minimized models will however needs further refinement in order to reduce the non-bonded interactions for the model to be judged as a good homology model.
3D Modeling of EMV2, LEA Protein from Vigna Radiata
Prediction of tertiary structure of a protein molecule signifies an important step towards understanding the structure–function relationships in the concerned protein family. Recently, the first solution structure of a LEA protein, LEA14 from Arabidopsis thaliana has been reported (Singh et al., 2005). In the present study, model of EMV2 LEA protein of Vigna radiata was generated from the LOOPP server, based on the structural homologues derived from the SCOP and protein data banks.
There exists biological sequence-structure deficit with more than 3 lakhs protein sequences and millions of partial nucleotide sequences, available in the public non-redundant databases (Boguski et al., 1994); and by contrast, the number of unique 3D structures in the protein data bank is still less than 1500 (Attwood and Parry-Smith, 2005). The difference of scale in sequence and structural information is an important factor to be considered when assigning functions to hypothetical proteins. Structure based functional implications of such proteins have always been speculative.
Generally, under stress situations the plants may induce formation of coiled coil / folding of the natively unfolded proteins into more rigid structures upon binding to the partner molecules. Since all natively unfolded proteins have defined partner molecules that can be as small as nucleotide or cations or a macromolecule, LEA proteins being natively unfolded is believed to have such binding partners to attain a rigid structures.
The LEA proteins from Vigna radiata are being classified as Group 1 LEA protein because of its extreme hydrophilicity and adoption of helical conformation as revealed by ab initio secondary structure predictions, in combination with the predominant random-coiled arrangement of the residues of Vigna LEA protein, is hypothesized to function as water replacement molecule. Such a property may facilitate hydrogen bonding of this EMV proteins with essentially any macromolecular or membrane surface. However, additional experiments on physico-chemical analyses including examination of hydration properties of these proteins need to be done to determine if EMV proteins can adopt certain structures upon interaction with other macromolecules.
Structure homologues identified for these EMV proteins show closest structural homology to proteins with helical bundles of small proteins and DNA/RNA binding proteins. These observations are contradictory to the earlier findings from our group that the low molecular weight protein isolated from Vigna was believed to be located in the cytoplasm The structural motifs of these proteins are predicted to form amphipathic a-helices which may be important for their function in protecting cells against dehydration. However, not all LEA proteins are folded and structured. Group 1 LEA proteins are reported to be very hydrophilic, loosely structured with predominantly random-coiled structures. These proteins are reported to form regular a-helical structure when subjected to altered physiological conditions. Observations from ab initio predictions of these Em proteins of Vigna indicate 32.32% of EMV2 proteins attains helical conformation as represented by helical blocks in the 3D hypothetical models (Manickam and Carlier, 1980).
Temperature-induced extended helix/random coil transition was reported for a Group 1 LEA protein from soybean. These proteins are by native, largely unstructured but attained 6- 14% helical conformation under temperature stress or at high salt concentrations (Soulages et al., 2002). Similar reports from (Goyal et al., 2003) for AavLEA1, a Group 3 LEA protein from the nematode, Aphelenchus avenae indicate oligomerization of these proteins in immunoblotting and cross-linking experiments, however majority of these proteins was found to be monomeric in analytical ultracentrifugation and gel filtration studies. Also, formation of a-helical structures on drying was reported in partially characterized protein from Typha latifolia, probably a Group 3 LEA protein based on Fourier transform-Infra red (FT-IR) spectroscopy studies (Wolkers et al., 2001).
The LEA proteins from Vigna radiata are being classified as Group 1 LEA protein because of its extreme hydrophilicity and adoption of helical conformation as revealed by ab initio secondary structure predictions, in combination with the predominant random-coiled arrangement of the residues of Vigna LEA protein, is hypothesized to function as water replacement molecule. Such a property may facilitate hydrogen bonding of this EMV proteins with essentially any macromolecular or membrane surface. However, additional experiments on physico-chemical analyses including examination of hydration properties of these proteins need to be done to determine if EMV proteins can adopt certain structures upon interaction with other macromolecules.
Validation of the Model
The hypothetical protein model generated was subjected to structure validation, for testing the accuracy of the model. The quality of the final ensemble of conformers was assessed using PROCHECK, a protein structure validation program. The visual displays of the models were performed with either the Swiss PDB viewer (Guex and Peitsch, 1997) or RasTop (Sayle and Milner-white, 1995).
Stereochemical parameters of the proteins like main-and side chains data of EMV2 was considered for determining the quality of the model. The main chain parameters like Ramachandran plot quality; peptide bond planarity, C-alpha chirality and over-all G factor are found to be within the limits for the model. However, the bad contacts per 100 residues are high. The side chain parameters are in better range and within the limits for EMV2. These parameters are compared to essentially satisfy the generated models with well-refined structures at similar resolution as described by (Morris et al., 1992).
The validation reports for the protein models are analyzed, and energy minimization of the models was made after checking the force field energy of the models. For a model to be validated based on quality, a good quality protein model should have 90% or more residues in the most favored regions of quadrangle in the Ramachandran plot. In the generated model of EMV2, the distribution of residues in the most favored regions is 87.6 and 93 %, respectively. This infers that EMV2 as good hypothetical protein model. Also, a tertiary structure of a protein can be worth considering only from its solution structures, obtained from the experimentations using either NMR or crystallographic studies. The homology model of mungbean LEA proteins, thus generated in this study, could some extent stimulate investigations at determining the mechanistic function of this stress associated proteins.
Functional implications based on the structural homologues hypothesize EMV2 to act as a water replacement molecule. LEA proteins generally are reported to posses multiple functions like salt, drought, heat and cold tolerance. Hence, the present study will be useful in further in vitro studies by over expression in model systems like E.coli or yeast cells and the recombinant protein can be subjected to Salinity, Cold shock, thermal stability analysis and the stress induced structural changes can be monitored to ascertain the possible functions of this important class of proteins.
S.Rajesh is grateful to the Council of Scientific and Industrial Research, New Delhi for grant of Research Fellowship. We are grateful to the crews of NCBI, EBI, MRC Lab-UK and SIB for making computational biology data/ tools publicly available.