Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 2
Research focusing on glutamate as a major contributor to schizophrenia has attained increasing prominence over the past decade. Analogous to an NMDA-receptor hypofunction in schizophrenia, growing evidence suggest that the disease is related to an excess of brain kynurenic acid (KYNA), an endogenous antagonist at the glycine-site of the NMDA receptor. Previous studies have shown tha t MK-801, an NMDA-receptor antagonist with psychotomimetic properties, induces alteration of several genes and protein levels in cortex and thalamus previously found to be changed in the brains of patients with schizophrenia. In the present study, we use p roteomics to investigate whether an increased KYNA turnover in the brain, induced by subchronic treatment of kynurenine and probenecid, w ould interfere with the protein synthesis in the cortex and thalamus in the rat brain. The levels of four proteins in the cortex wer e increased in the group treated with kynurenine and probenecid compared to vehicle-treated controls. The proteins were; 1, Ubiquitin carbo xy- terminal hydrolase L1 (UCHL1), 2, Similar to NADH dehydrogenase, 3, Cytochrome c oxidase and 4, protein with an undetermined identity. No protein changes were observed in the thalamus. Two of these proteins are implicated in mitochondrial energy produc tions and mRNA from one of them – cytochrome c oxidase – has previously been shown to be increased in the cortex from patients with schizophrenia. Present result show that increased turnover of the endogenous NMDA receptor antagonist KYNA is able to affect cortical protein synthesis to a condition as observed in patients with schizophrenia.
Keywords: kynurenic acid, Probenecid, Two-dimensional gel electrophoresis, Rat model, Proteomics, Schizophrenia
The underlying cause of schizophrenia has traditionally been attributed to dopaminergic hyperactivity in the brain (Abi-Dargham et al., 2000); (Carlsson and Lindqvist, 1963). Over the past decade though, research focusing on glutamate as a major contributor to the disease has attained increasing prominence (Carlsson et al., 2001; Javitt, 2004; Javitt and Zukin, 1991; Jentsch and Roth, 1999; Kim et al., 1980; McCullumsmith et al., 2004; Coyle, 2004; Coyle, 2006). An NMDA-receptor hypofunction in schizophrenia comes above all from clinical observations that NMDA-receptor antagonists like phencyclidine (PCP) and ketamine induce schizophrenia-like symptoms including both positive and negative symptoms as well as cognitive deficits (Adler et al., 1999; Itil et al., 1967; Luby, 1959). Thus, a dysregulation of dopamine (DA) transmission in schizophrenia might be secondary to alterations in glutamatergic N-methyl-D-aspartate (NMDA)-receptor mediated transmission (Carlsson et al., 2004; Grace, 1991; Olney and Farber, 1995). Analogous to an NMDA-receptor hypofunction in schizophrenia, growing evidence suggest that the disease is related to an excess of brain kynurenic acid (KYNA), an endogenous antagonist at the glycine-site of the NMDA receptor. Thus, KYNA is elevated in the cerebrospinal fluid (CSF) (Erhardt et al., 2001a; Nilsson et al., 2005) as well as in the post-mortem brain (Schwarcz et al., 2001) in patients with schizophrenia. Furthermore, preclinical studies suggest that the compound tonically modulates the impulse activity of DA neurons in the ventral tegmental area (Erhardt and Engberg, 2002; Nilsson et al., 2006; Schwieler et al., 2006) and causes disruption of prepulse inhibition (Erhardt et al., 2004), a behavioral model of schizophrenia.
Proteome analyses may serve as a useful strategy allowing for identification of molecular mechanisms underlying the pathophysiology of schizophrenia. Previous studies have shown that MK- 801, an NMDA-receptor antagonist, induces alterations in cortical and thalamic levels of several genes and proteins previously found to be changed in the post-mortem brain from patients with schizophrenia (Paulson et al., 2004a; Paulson et al., 2004b; Paulson et al., 2003). Therefore, we have investigated whether increased KYNA turnover in the brain (Nilsson et al., 2006), induced by subchronic treatment with kynurenine, the precursor of KYNA, and probenecid which prevents the efflux of KYNA from the brain, would interfere with protein synthesis in the cortex and the thalamus in the rat.
Subjects
Male Sprague-Dawley rats (Scanbur BK, Sollentuna, Sweden; weighing a minimum of 180 g (at day of surgery) and a maximum of 350 g (at day of experiment)) were housed in groups of three or four with ad libitum feeding. Environmental conditions were checked daily and maintained under constant temperature (25°C), and 40-60% humidity in a room with a regulated 12-h light/dark cycle (lights on at 06.00 AM, off at 06.00 PM). Experiments were approved by and performed in accordance with the guidelines of the Ethical Committee of Northern Stockholm, Sweden, and all efforts were made to minimize the number of animals used and their suffering. To subchronically elevate endogenous brain KYNA concentration, rats were administered kynurenine and probenecid for 14 days via osmotic pumps with a continuous flow of 5 μl/h. Osmotic pumps (2ML2 Alzet, USA) were filled under aseptic conditions with either vehicle (0.1 M Tris in deionised water; pH adjusted to 7.4 with acetic acid), kynurenine (dissolved in deionised water; pH adjusted to 3.5 with NaHCO3) in a concentration equivalent to approximately 20 mg/kg/day at day of surgery, or probenecid (dissolved in 0.1 M Tris buffer; pH 8.0) in a concentration equivalent to approximately 10 mg/kg/day at the day of surgery. Both drugs were dissolved in the highest possible concentration permitted in the osmotic pumps (volume: 2 mL) and all solutions were filtered through a sterile filter (Acrodisc Syringe Filter 0.2 μm Supor Membrane) before filling of pumps. The osmotic pumps were inserted through an incision in the neck and placed subcutaneously on the back of the rats during chloral hydrate anaesthesia (400 mg/kg, i.p.). After surgery the rats were placed in single cages to awake for 24 hours before reunited in groups of 3- 4 per cage. After 14 days, electrophysiological or behavioral experiments were performed. Immediately after each experiment the rats were killed by decapitation. The right hemisphere was used for HPLC analyses of KYNA (see Nilsson et al., 2006). Electrophysiological, behavioral and KYNA data from these rats have previously been published (Nilsson et al., 2006).The remaining hemisphere was placed on an ice-cooled metal surface and the thalamus and cerebral cortex were dissected. Each tissue sample was put in an ice-cooled Eppendorf tube and frozen on dry ice before storage at –80°C in a freezer for later proteome analysis.
Two-dimensional Gel Electrophoresis
Proteome analyses were performed on the cortex and thalamus using two-dimensional gel electrophoresis (2-DGE) and mass spectrometry (MS). For analysis of quantitative differences, approximately 100 protein spots were taken into account in the proteomic analysis of cortex and approximately 200 protein spots in thalamus. The thalamus and cortex, approx. 25-50 mg wet weight of each, were extracted as previously described (Paulson et al., 2004b). The protein sample (30 μL, 300 μg) was mixed with 160 mL rehydration buffer (9 M urea, 4 % immobilized pH gradient (IPG) buffer, bromphenolblue) and 160 mL isobuffer (9 M urea, 65 mM 3-[(3- cholamidopropyl)-dimethylammonio]-1-propanosulfonate hydrate (CHAPS), 35 mM tris, 65 mM dithiothreitol (DTT), bromphenolblue). To separate the proteins 2-DGE was performed. In the first dimension Ready StripTM IPG strips, pH 5-8, 11 cm (BioRad) were used in a Protean IEF Cell (BioRad). The second dimension was carried out using 12% Criterion XT Bis-Tris Gels (BioRad) in a Criterion Dodeca Cell (BioRad) combined with 3-[Nmorpholino] propane sulfonic acid (MOPS) running buffer (50 m M MOPS, 50 mM tris, 3.5 mM SDS, 0.8 mM EDTA ) at a constant voltage (200 V), for 60 min. The gels were stained with SYPRO Ruby Protein Stain (Molecular Probes, Eugene, OR, USA) according to the supplier’s protocol. Image acquisition and analysis were performed on a LAS-3000 (Fuji). The protein spots were detected, quantified and matched using the PD-Quest 2D-gel analysis software, 7.4. The gels were normalized according to the total protein density of detected spots in each gel. Only proteins with significantly altered levels (Mann-Whitney p<0.05) in the kynurenine- and probenecid -treated rats as compared to vehicle treated rats were reported. Altered proteins were excised for identification using MS.
In-gel Protein Digestion
The gel spots with significantly up or down regulated intensity were excised from the SYPRO-stained 2-D gels and enzymatic cleaved with trypsin as described previously (Paulson et al., 2004b), with some modifications. Briefly, the gel pieces were washed in 100 μL 1:1 H2O:acetonitrile (ACN) 2 x 15 min and then destained and dried with 50 μL ACN. The gel pieces were rehydrated in 10 μL chilled digestion buffer (50 mM NH4CO3, 12.5 ng/μL trypsin) at 37°C overnight. The supernatant was collected and the peptides extracted twice with 30μL 5% formic acid (FA): ACN (1:1). The combined supernatants were lyophilized and dissolved in 10 μL 0.1% formic acid (FA), prior to analysis.
Mass Spectrometry and Data Base Searches
Mass analysis of protein digests were performed in reflectron mode with a MALDI-TOF MS (Autoflex, Bruker-Franzen Analytik GmbH, Germany). A stainless steel MALDI target 400/384 TF (AnchorChipTM, Bruker Daltonik, Bremen, Germany) with circular interruptions, which act as hydrophilic sample anchors, was used (Schuerenberg et al., 2000). A thin layer of α-cyano-4-hydroxycinnamic acid (CHCA; 100 g/L CHCA in 90% acetone, 0.005% TFA (v/v)) crystals was spread out on all the anchors on the sample plate. 1 μL of each sample was deposited onto an anchor point. After two minutes the remaining liquid was removed and the target was washed by immersing it in a solution of 0.1% trifluoric acid (TFA) for 10 seconds. MS spectra (Figure 2) were processed using Flex-analysis (Bruker) and used without further interpretation for database searches against all entries in the NCBI nr database with MASCOT (http:// www.matrixscience.com). A mass deviation of 50 ppm was used, and one missed cleavage and Rattus norvegicus were specified. Only those protein identities obtained with >95% confidences using MALDI-TOF MS were considered.

Figure 2: Mass spectra of a) cytochrome c oxidase, b) Ubiquitin carboxy-terminal hydrolase L1, c) NADH dehydrogenase, and d) unidentified protein.
Proteome analyses were performed in the cortex and thalamus using 2-DGE and MS. For analysis of quantitative differences, approximately 100 protein spots were taken into account in the proteomic analysis of the cortex and approximately 200 protein spots in the thalamus. In cortex four protein levels were increased with >95% significance in the group treated with kynurenine and probenecide (n=11) compared to saline controls (n=15). The proteins were; ubiquitin carboxy-terminal hydrolase L1 (UCHL1), Similar to NADH dehydrogenase, cytochrome c oxidase and a fourth protein that could not be positively identified. This protein has a pI of ~5.5 and a MW of ~10. A summary of the altered protein levels in cortex is presented in Table 1. Figure 1 shows a typical SYPRO Ruby stained 2D-gel of the cortex proteins in kynurenine and probenecid treated rats, with numbers indicating altered and excised protein spots. No changes were observed in the thalamus.
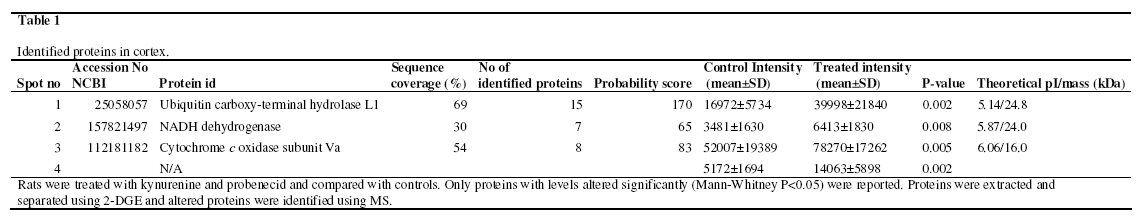

Figure 1: A typical SYPRO Ruby stained 2D-gels of cortex proteins in kynurenine and probenecid treated rats, with numbers indicating altered and excised protein spots. The proteins were separated by 2-DGE, quantified by PD-Quest software and identified by MS as described in Methods. Only the protein levels that were significantly altered (Mann-Whitney p<0.05) are reported. Protein identities obtained with > 95 % confidences by MALDI TOF-MS were considered. Proteins with altered levels are indicated corresponding to Table 1.
In the present study, kynurenine and probenecid was administered subchronically in order to increase brain KYNA turnover, thereby mimicking a situation of hypoglutamatergia and hyperdopaminergia as proposed in schizophrenia (see Introduction). This model was used to screen for aberrations of the proteome in rat thalamus and cortex in order to validate and increase the understanding of the kynurenic acid hypothesis of schizophrenia. The finding of the present study is that subchronic treatment with kynurenine and probenecid is associated with altered rat cortical levels of the proteins UCHL1, Similar to NADH dehydrogenase, and cytochrome c oxidase. In agreement with the present results, cytochrome c oxidase gene expression has previously been reported to be upregulated in rats subchronically treated with the NMDA receptor antagonist MK-801 (Paulson et al., 2004a; Paulson et al., 2004b; Paulson et al., 2003)
Several lines of evidence indicate that schizophrenia is associated with changes in mitochondrial energy production in the brain (Ben-Shachar, 2002). Traditionally, peptides such as cytochrome c oxidase, a key enzyme in the respiratory chain producing metabolic energy, and NADH dehydrogenase has been used as markers in reflecting neuronal energy metabolism and neuronal function in general (Prince et al., 1999). The first paper reporting an involvement of oxidative metabolism in schizophrenia was published in the mid 1950’s (Takahashi et al., 1954; see Maurer et al., 2001) and showed lowered aerobic glycolysis in patients with schizophrenia. Although this original finding has been replicated, the picture is probably more complex - in many studies the results might have been confounded by medication effects, chronic illness and difficulties of measurement (Andreasen et al., 1997). More recent studies have found both decreased as well as increased metabolic activity in patients with schizophrenia which may be explained by an imbalance in cortical and subcortical circuits (Andreasen et al., 1997). In agreement with present data, Mulcrone and collegues (Mulcrone et al., 1995) have shown that the mRNA of cytochrome c oxidase is increased in the cortex in patients with schizophrenia, tentatively reflecting increased energy metabolism.
The finding that the levels of Similar to NADH dehydrogenase, and cytochrome c oxidase was increased in the present study, suggest per se that elevated levels of brain KYNA increase brain energy metabolism in the rat. In a recent study we reported that enhanced turnover of KYNA, as induced by using the present protocol, increase neuronal firing of VTA DA neurons (Nilsson et al., 2006). Clearly, neuronal activity demands high energy consumption and there is a fine-tuned coupling between firing rate and mitochondrial function of a neuron (see Kann & Kovacs, 2007). Indeed, PCP and MK-801 have been shown to produce EEG changes with high-amplitude cortical activity (Marquis et al., 1989). Recent studies from our laboratory are in consonance with this observation. Thus, acutely or subchronically elevated levels of brain KYNA increase the firing of rat midbrain DA neurons (Erhardt and Engberg, 2002; Erhardt et al., 2001b; Nilsson et al., 2006; Schwieler et al., 2006; Linderholm et al., 2007) and disrupt PPI in rats (Erhardt et al., 2004). These findings are supported by clinical studies showing that CSF KYNA positively correlates to CSF homovanillic acid in healthy controls as well as in patients with schizophrenia, suggesting that increased brain KYNA is associated with an increased turnover of DA (Nilsson et al., 2007a; Nilsson et al., 2007b).
UCHL1 is an abundant protein making up 2% of all proteins in the brain (Wilkinson et al., 1989). It is responsible for hydrolysis of polyubiquitin chains into monomeric ubiquitin and belongs to a family of deubiquitinating enzymes (Pickart, 2000). Mutations in the gene encoding for UCHL1 result in a 50% decrease of catalytic activity, implying that increase of UCHL1 activity might lead to increased ubiquitination and therefore enhanced clearance of abnormal proteins. It has been suggested that UCHL1 plays an essential role in the pathogenesis of neurodegenerative disorders (Ross and Pickart, 2004). The significance of our finding that UCHL1 is increased following elevated KYNA turnover is obscure but tentatively this protein may, at least to some extent, participate in the well-known neuroprotective actions of KYNA (see Stone, 2000).
Following 2 weeks of kynurenine and probenecid administration, electrophysiological and behavioural experiments along with analysis of brain and blood levels of KYNA was performed. This treatment enhanced dopaminergic firing activity and tended to disrupt PPI (Nillson et al., 2006). However, whereas a single dose of kynurenine and probenecid (corresponding to a daily dose of the subchronic treatment) as well as a four-day treatment with the compounds significantly increased brain KYNA concentration, the subchronic treatment (14 days) did not produce elevated whole brain levels (Nilsson et al., 2006). The lack of increase in whole brain KYNA levels at day 14 may per se point to a development of tolerance in the conversion of kynurenine into KYNA with subchronic kynurenine and probenecid treatment. However, this appears unlikely since the subchronic treatment produced effects on spontaneous VTA DA cell firing identical in magnitude to those observed following acute elevation of brain KYNA (Erhardt and Engberg, 2002; Nilsson et al., 2006; Schwieler et al., 2006; Linderholm et al., 2007). Rather, the present effects of subchronic treatment with kynurenine and probenecid should be related to an increased turnover of KYNA involving increased release to, and elimination from glutamatergic boutons (Curatolo et al., 1996; Guillemin et al., 2001; Kiss et al., 2003; Swartz et al., 1990) enough for possible receptor interaction (Turski et al., 1989), but without producing a detectable increase in whole brain KYNA concentration. In this regard the present results are in harmony with previous findings where e.g. subchronic L-DOPA treatment is found to produce motoric sensitisation in spite of the lack of a striatal DA elevation (Carey, 1991; Carey, 1993).
Moreover, kynurenine is the precursor of several kynurenines, e.g. quinolinic acid, an excitotoxic NMDA-receptor agonist (Stone and Perkins, 1981), and we cannot exclude the possibily that other kynurenines than KYNA are responsible for the present observations. Indeed, probenecid, which was given to prevent the efflux of KYNA out of the brain (Moroni et al., 1988), would also increase quinolinic acid, which is extruded via the same probenecidsensitive carrier (Morrison et al., 1999). However, in support of a prevailing role of KYNA in this regard is the fact that administration of kynurenine, alone or in combination with probenecid, display anticonvulsant effects and attenuates quinolinic acid induced neurotoxicity in rats (Nozaki and Beal, 1992; Santamaria et al., 1996; Vecsei et al., 1992). Moreover, subchronic administration of kynurenine and probenecid was previously found to be associated with increased neuronal activity of VTA DA neurons (Nilsson et al., 2006), effects also observed following acute pharmacological elevation of KYNA (Erhardt and Engberg, 2002). These effects of elevated KYNA levels have previously been demonstrated to be executed via blockade of the NMDA receptor (Erhardt and Engberg, 2002; Erhardt et al., 2002; Linderholm et al., 2007), thus strongly arguing against a significant role of quinolinic acid in the present study. Since schizophrenia is associated with a dysfunction of dopaminergic systems tentatively induced by increased levels of brain KYNA (Erhardt et al., 2001; Schwarcz et al., 2001; Nilsson et al., 2005), novel treatment of the disease could rationally be directed towards brain KYNA formation. The development of specific kynurenine aminotransferase (KAT) II inhibitors (Pellicciari et al., 2006) that decrease brain KYNA concentrations could thus be of importance in the treatment of schizophrenia. In support of this notion, cyclooxygenase (COX)-2 inhibitors (which reduce rat brain KYNA levels as well as decrease midbrain dopaminergic activity; Schwieler et al., 2005; Schwieler et al., 2006) added to conventional antipsychotic treatment, display beneficial effects with regard to both positive and negative symptoms in patients with schizophrenia (Müller et al., 2002; Müller et al., 2004).
It would be interesting to explore protein levels in rats following a ro bust and prolonged elevation of brain KYNA levels. In the present paper, no protein levels in the thalamus and only four protein levels in the cortex were changed following subchronic blockade of NMDA receptors, differences tentatively explained by the lack of confirmed increase in KYNA levels. In our previous papers, changes in both thalamus and cortex have been observed and several more proteins are altered (Paulson et al., 2003; Paulson et al., 2004a; Paulson et al.,2004b). For example both the levels of glutamate decarboxylase (GAD) and the levels of the GABA transporter (GAT) were found to be altered in the rat frontal cortex following chronic administration of MK-801 (Paulson et al., 2003). In addition, it has been suggested that hypofunction of the NMDA receptor, tentatively caused by elevated levels of endogenous KYNA, induces GABAergic dysfunction in schizophrenia (Benes and Berretta, 2001; Coyle, 2004; Coyle and Tsai, 2004). In follow-up studies, the most important and interesting proteins to explore following a robust and prolonged elevation of brain KYNA levels would therefore be the levels of GAD and GAT.
In conclusion, the present study shows that subchronic treatment with kynurenine and probenecid results in increased cortical levels of four proteins. Two of these proteins are implicated in mitochondrial energy productions and mRNA from one of them – cytochrom C oxidase - is increased in the cortex from patients with schizophrenia. Present result show that increased turnover of the endogenous NMDA receptor antagonist KYNA is able to affect cortical protein synthesis to a condition as observed in patients with schizophrenia.
The authors would like to dedicate this paper to Professor Peter S. Eriksson who passed away during the final preparation of this paper. This study was supported by Hållstens Forskningsstiftelse, the Swedish Brain Foundation, the Swedish Medical Research Council (K2006-21X-07484-21-3 and 529-2004-6488), Svenska Läkaresällskapet, Torsten och Ragnar Söderbergs Stiftelser and the Karolinska Institutet.
It was assumed that each data set analysed is only a homogenous part of the total proteome of a given species. Then the fitted DEL model formula and the hypothetical distribution of the total population of proteins of a given organism (see Appendix 2) are related in the proportion:
nK def a1 exp( -d1k ) + a2 exp ( -d2k ) NP
== (A1.1)
nk* a1* exp( -d1k ) + a2* exp (-d2k)Np*
where a1* and a2* are the amplitudes of a hypothetical distribution for the total population, Np is the extrapolated size of the analysed probe and Np* is the total size of proteome.
In the above ratio, Np value includes interacting proteins ( Nk>0 ) and also non-interacting ones ( n0 ) - not included in the investigated data sets, so that:
Np = n0 + Nk>0 (A.1.2)
As eq. A.1.1 is fulfilled for each node degree and for different decay constants d1 and d2 , it should be:
a1* = a1 /sc (A.1.3)
a2* = a2 / sc (A.1.4)
where the scaling factor equals to:
n0 + Nk>0
sc= (A.1.5)
Np*
Let us consider protein interaction network containing two classes of proteins (namely 1 and 2) characterized by different dynamics of evolutional performance, i.e., emerging with the rates q1 and q2 (as non-interacting at the beginning), then gaining some interactions with the rates ν1 and ν2 , and being eliminated with the rates γ1 and γ2 - per protein. All mentioned rates are assumed as being distinct and constant.
A number of selected proteins of a given class δNi*(i=1,2), originated within small period of time , vanishes with age a according the equation
dδNi*
= - γiΔNi* i = 1, 2 (A.2.1)
da
with an initial condition
Ni*|a=0 = qiδt i= 1, 2 (A.2.2)
The resolution of eqs. A.2.1 and A.2.2 represents the exponentially diminishing course
δNi* = qiδt exp( - γia) i= 1, 2 (A.2.3)
The assumed continuous approximation and linear increase in protein connectivity
K = νia (A.2.4)
and also the relationship , let us to transform eq. A.2.3 into the formula
qi γi
δNi*= δK exp( - K ) i=1, 2 (A.2.5)
νiνi
which integrated within successive intervals [k, k+1] indicates the number of k-degree proteins of class "i" , , equal to
qi γiγi
nki*= (1 -exp( - ) ) exp( - K ) i=1, 2 (A.2.6)
γi νi νi
Now, the total distribution of node degree, nk*, where nk* = nk1* + nk2*, may be written in the double-exponential form:
nk* = a1* exp( -d1k ) + a2* exp( -d2k ) (A.2.7)
The symbols introduced above mean
q1 γ1
a1*= (1 -exp( - ) ) (A.2.8)
γ1 ν1
q2 γ2
a2*= (1 -exp( - ) ) (A.2.9)
γ2 ν2
γ1
d1 = (A.2.10)
ν1
γ2
d2 = (A.2.11)
ν2
A contribution of "i " class proteins in eqs. A.2.7 formally vanishes for K > ζevi −1, where is the time of evolution of interactome. Thus the index k should not exceed max[ ζev1 −1, ζev2 −1 ] Assuming a relatively high value ζe ( >> 1/ νi ), by summation of a superposition of geometrical series nk*described by the eq. A.2.7 over 0 ≤ k ≤ ∞ , one can obtain the total size of proteome : Np*
q1 q 2
Np*= + (A.2.12)
γ1 γ2
with a distinguished levels of class contribution
q1
N1*= (A.2.13)
γ1
and
q2
N2*= (A.2.14)
Y2