Journal of Probiotics & Health
Open Access
ISSN: 2329-8901
ISSN: 2329-8901
Research Article - (2022)Volume 10, Issue 8
Mango is a delicious and nutritious fruit widely distributed in tropical and subtropical countries and regions. In China, especially in Baise, mango is one of the most famous fruits because of its tasting. In the local agricultural income, the mango industry occupies an extremely important position. Therefore, the research on mangoes and the prevention and treatment of their root diseases are directly related to the local economic development. More and more studies have shown that microbiota plays an important role in crop plant growth and disease prevention, and contributes to the sustainable development of agriculture in general. This study used high-throughput sequencing technology to study the soil microbial diversity of mango roots in Baise. The results showed that the soil where the roots grow normally and the soil with diseased roots have a certain degree of similarity and differences. The main types of bacterial communities are Proteobacteria, Acidobacteria, Verrucomicrobia, Planctomveetes and Bacteroidetes. Compared with normal soil, the relative abundance of Proteobacteria, Verrucomicrobia and Gemmatimonadetes is low, and the relative abundance of Acidobacteria, Bacteroidetes, Bacteroidota, Planctomveetes and Rookubacteria is high. The analysis of the difference between the Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways showed that the two groups of samples had a higher abundance ratio and significant differences in functions such as amino acid metabolism, metabolism of cofactors and vitamins (P<0.05). Clusters of Orthologous Groups (COG) analysis results showed that in addition to extracellular structure, RNA processing and modification, and cytoskeleton function, other functions were significantly different in the two samples (P<0.05). These results revealed for the first time the changes in soil microbial diversity between normal and diseased roots of mangoes, and provided a theoretical basis for guiding the healthy planting and biological control of the root diseases of mangoes.
Mango; Soil microbial diversity; Disease control; Significant differences; Gemmatimonadetes
Mango (Mangifera indica Linn.) belongs to the genus Mango in the Aceraceae family. It is an evergreen tree widely distributed in tropical and subtropical countries and regions. There are about 200 mango varieties or strains preserved in China, mainly distributed in China's Guangxi, Hainan, Yunnan, Guangdong, Fujian and other provinces (regions) [1]. The total cultivated area and total output of mangoes in Guangxi both rank top 1 in the country. Mango cultivation is mainly distributed in Youjiang District, Tianyang, Tiandong and other places, especially in Youjiang District of Baise City. In 2018, the output value of mangoes was 850 million yuan.
Mango is delicious and rich in nutrients. It is an important source of a various nutrients such as carbohydrates, fats and fatty acids, proteins and amino acids, and organic acids. In addition, mangoes also contain micronutrients, such as vitamins and minerals [2]. As one of many specialty fruits in Guangxi, it has attracted much research attention because of its remarkable economic benefits. However, these studies most focused on mango diseases and insect pests, and there were few reports on mango plants, especially their rhizosphere diseases. Plants infected by soil-borne pathogens will suffer root rot, root growth inhibition, growth retardation; stem rot, wilting and even plant death [3,4]. The rhizosphere is a key area where plants directly affect the soil around the roots. It is a hot spot for microbial community activities, such as microbial interaction and gene exchange [5]. There is a close two-way interaction between the microbial community and the plant host, which is an important determinant of plant health and productivity [6]. More and more studies have shown that microbiota plays an important role in crop plant growth and resistance to environmental stress, because certain beneficial microorganisms can promote plant growth and stress resistance [7-10]. With the advancement of analysis technology, scholars have carried out research to explore the interrelationship between plants and microorganisms, and to understand the relevant mechanisms of increasing crop yields [11,12]. If the characteristics of microbial communities formed in the rhizosphere and their effects on plants are analyzed, these characteristics can be used as possible sustainable substitutes in agricultural planting to improve stability and long-term crop productivity [13,14]. It has been difficult to study these complex interactions at the community level. However, high-throughput sequencing methods have recently been used to prove the resistance of tomato rhizosphere microorganisms to Ralstonia. It has been found that the relative abundance of Flavobacterium in the root system of disease-resistant species is far higher than the susceptible species. The Flavobacterium TRG1 obtained by splicing and assembling subsequently improved the resistance of tomato to bacterial wilt [15].
In this study, in a mango orchard in Baise, Guangxi, it was found that the plants with infected roots had shorter plants and reduced yields than normal plants. Two kinds of root soil samples were collected and analyzed using high-throughput technology. The structure of soil microbial community was studied. The root causes of plant disease were explored from the perspective of root microbial community while providing a theoretical basis for healthy planting of mango plants.
Site and soil collection
Soil as a microbial inoculum was collected from Leye Agricultural Investment Co., Ltd Farm (18°644′N, 108°760′E) in Baise city, Guangxi, China. Covering vegetation was stripped off and rhizosphere soil from 10 years old mango trees was collected from a depth of 15 cm. Healthy and intact root samples (5-8 cm in length) with similar diameters were cut and stored in sterile sample bags to avoid contaminations from other foreign bacteria.
DNA extraction
The DNA extraction processes of soil samples were carried out strictly following the manufacturer’s instructions. Before DNA extraction, the soil samples were frozen in liquid nitrogen. DNA was extracted from 0.25 gm of each soil sample using E.Z.N.A. soil DNA kits (OMEGA, United States) following the manufacturer’s instructions. After thawing on ice, DNA quality was determined by Polymerase Chain Reaction (PCR) amplification and electrophoresis using a 0.8% agarose gel. Sample quality was determined using a NanoDrop instrument (NanoDrop Technologies, Inc., Wilmington, DE, USA).
Soil physicochemical analyses
Soil pH was determined using 1 M potassium chloride (KCl) (2:5-soil to solution ratio) solution with IQ Scientific 150 pH meter (Spectrum Technologies, Plainfield, IL, USA)(Kome, et al.).
Data analysis
Third generation microbial diversity was performed at Beijing Biomarker Technologies. The soil DNA of 12 plants was divided into two groups, each with 6 DNA. Third generation microbial diversity was obtained by analysis of sequencing on Pacbio sequencing platform. SMRT Cell method was used to sequence marker genes. Then Circular Consensus Sequencing (CCS) was filtered to get Optimization-CCS for Operational Taxonomic Units (OTUs) clustering. In this study, microbial diversity is mainly based on the conserved region of nucleic acid.
Quantification of gene expressions by real-time PCR
Third generation microbial diversity was performed at Beijing Biomarker Technologies. The soil DNA of 12 plants was divided into two groups, each with 6 DNA. Third generation microbial diversity was obtained by analysis of sequencing on Pacbio sequencing platform. Single Molecule Real Time (SMRT) sequencing cell method was used to sequence marker genes. Then Circular Consensus Sequencing (CCS) was filtered to get optimization-CCS for Operational Taxonomic Units (OTUs) clustering. In this study, microbial diversity is mainly based on the conserved region of nucleic acid sequences that encode ribosomal RNA. For bacteria it will be 16S region. In these sequences, there are both conserved regions and variable regions. The conserved regions reflect the relationship between biological species, while the hypervariable regions reflect the difference between species. Based on OTU analysis results, taxonomic analysis was performed on samples at various taxonomic levels to obtain the community structure map, species cluster heat map, genus level phylogenetic tree and taxonomic tree of each sample at the taxonomic level of phylum, class, order, family, genus and species. The species diversity within a single sample was studied using Alpha diversity analysis. Ace, Chao1, Shannon and Simpson indexes of each sample were statistically calculated at 97% similarity level to draw sample dilution curve and rank abundance curve. Use 16S functional gene prediction analysis to predict genes’ function and calculate the richness of functional genesis for samples.
Soil physicochemical analyses
The pH of the diseased sample is 5.6, and the pH of the normal sample is 6.0 (Figures 1A and 1B).

Figure 1: Soil physicochemical analysis of mango plants. (A): The normal M1; (B): The diseased mango BM1.
CCS sequences and OTU statistics
In this study, the diseased soil samples collected in the mango orchard were labeled BM1, and the normal soil samples were labeled M1. After two samples were sequenced and identified by Barcode, a total of 14985 CCS sequences were obtained. The diseased samples had 7501 CCS sequences, and the normal samples had 7484 CCS sequences.
In order to investigate the difference in microbial community structures between diseased soil and normal soil, we selected two diseased and normal soil samples from mango fields for high-throughput sequencing to analyze the microbial community structure of rhizosphere soil. Among them, a certain number of sequences are randomly selected from the samples, and a dilution curve is constructed based on the number of sequences extracted and the number of OTUs which can be used to determine whether the amount of sequencing for each sample is sufficient. Judging from the trends in Figures 2A and 2B, the rarefaction curves of the two samples with the continuous increase in sequencing volume, the increase in the number of OTUs gradually tended to be flat, but still did not reach saturation, indicating that there are still a small number of new microbial species be found. However, the Shannon index of each sample is saturated, and the curve is basically flat, indicating that the sequencing depth is large enough and the amount of data obtained can fully reflect the species and classification information in each sample (Figure 2A).

Figure 2: Curves of the samples. (A): Multi sample rarefaction curves; (B): Multi sample shannon curves.
Microbial diversity analysis
Using QIIME2 software, the Alpha diversity index of the two soil samples collected from the Baise Mango orchard was evaluated. The Alpha diversity index value statistics of each sample are shown in Table 1. In terms of soil bacterial richness, the Accumulated Cyclone Energy (ACE) index/Chao1 index of diseased soil samples and normal soil samples are 550.736/532.9873 and 548.3214/524.987, respectively, and there is not much difference between the two. The diseased soil sample is higher than the normal soil sample. Bacterial soil diversity also showed that the diversity of diseased soil samples was higher than that of normal soil samples, with Shannon indexes of 7.940 and 7.373. The abundance of bacterial communities in diseased soil samples was slightly higher than that in normal soil samples. Table 1 shows that after the disease, the richness and diversity in the soil have become higher (Table 1).
| Sample ID | Feature | ACE | Chao1 | Simpson | Shannon | PD whole tree | Coverage |
|---|---|---|---|---|---|---|---|
| BM1 | 487 | 550.7358 | 548.3214 | 0.9921 | 7.9397 | 32.8597 | 0.9689 |
| M1 | 472 | 532.5828 | 524.9873 | 0.9834 | 7.3732 | 29.0978 | 0.9821 |
Note: PD: Phylogenetic Diversity; ACE: Accumulated Cyclone Energy
Table 1: Alpha diversity index statistics of each mango sample.
According to the analysis of the OTU Venn diagram as shown in Figure 3, the number of OTUs shared by the two soil samples is 268, indicating that the bacterial diversity between the two soil samples has a certain similarity. However, the number of unique features of the diseased sample is 219, and the normal sample has 204, indicating that there are differences between the two samples (Figure 3).

Figure 3: Operational Taxonomic Units (OTUs) venn diagram of the sample.
Microbial taxonomy composition analysis
Figures 4 and 5 respectively show the histograms of the species distribution of bacteria at the taxonomic level of the phylum and genus in the two soil samples. The histogram only shows the top 10 abundance species at each taxonomic level. A color represents a species, and the length of the color block represents the relative abundance ratio of the species. As can be seen from Figure 4, Proteobacteria, Acidobacteria, Verrucomicrobia, Planctomveetes and Bacteroidetes were the main bacteria groups in the two soil samples of Baise Mango Field. Compared with normal soil samples, the abundance of Proteobacteria, Verrucomicrobia and Gemmatimonadetes decreased, while the abundance of Acidobacteria, Bacteroidetes, Bacteroidota, Planctomveetes and Rookubacteria increased (Figures 4 and 5).

Figure 4: Distribution of species at phylum level.
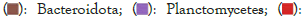
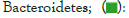
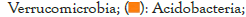


Figure 5: Distribution map of genus horizontal species.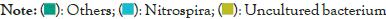

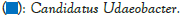
The dominant genus groups are mainly Candidatus Udaeobacter, Sphingomonas, uncultured bacterium Chitinophagaceae, and uncultured bacterium subgroup 6, uncultured bacterium WD2101 soil group. Candidatus Udaeobacter and Sphingomonas have higher abundance in normal samples. Uncultured bacterium Chitinophagaceae, uncultured bacterium Subgroup 6, uncultured bacterium WD2101 soil group, uncultured bacterium SC-I-84, Uncultured bacterium Rokubacteriales, Flavobacterium, etc. are more abundant in diseased samples.
Analysis of the composition and difference of metabolic pathways
For the functional abundance between different samples, use the G-TEST (large sample: the number of annotated functional genes is greater than 20) and Fisher (small sample: the number of annotated functional genes is less than 20) test methods in STAMP to perform a pair of samples. The significant difference test is performed on the pairwise T-test test between different groups, and the P-value threshold is 0.05 (<0.05 means significant).
The analysis of KEGG metabolic pathway differences between groups in Figure 6 shows that the two groups of samples have high abundance ratios in functions such as global and overview maps, amino acid metabolism, metabolism of cofactors and vitamins, energy metabolism, and carbohydrate metabolism. The difference ratio of functional abundance and p-value in the 95% confidence interval show that these functional differences of the two samples are significant. Microbial function was predicted and enumerated by PICRUSt2 and STAMP, it was found that the abundance expression of biosynthesis of other secondary metabolites, glycan biosynthesis and metabolism, translation, global and overview maps, folding, sorting and degradation, replication and repair, nucleotide metabolism, digestive system, metabolism of cofactors and vitamins, transport and catabolism, energy metabolism, cell motility and other functional genes in BM1 sample were significantly higher than those of M1 sample (P<0.05). However, the participation in endocrine system, xenobiotics biodegradation and metabolism, metabolism of other amino acids, membrane transport, lipid metabolism, amino acid metabolism, aging, etc. is significantly lower than M1 sample (P<0.05). By analyzing the composition and differences of KEGG metabolic pathways, it is possible to observe the differences and changes in the metabolic pathways of the functional genes of the microbial community between samples of different groups. This is an effective means to study the changes in the metabolic function of the community samples to adapt to environmental changes (Figure 6).

Figure 6: Analysis of differences in KEGG metabolic pathways between groups. Note: The picture shows the difference analysis diagram of the KEGG metabolic pathway under the second level. Different colors in the picture represent different groups. The left figure in the picture shows the abundance ratio of different functions in two samples or two groups of samples, the middle shows the difference ratio of the function abundance within the 95% confidence interval, and the rightmost value is the p value.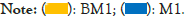
Clusters of Orthologous Groups (COG) of proteins are a functional classification database of proteins commonly used in prokaryotes. COG function prediction analysis method is basically the same as KEGG. The analysis results are shown in Figure 7. Except for extracellular structures, RNA processing and modification, and cytoskeleton functions, the other functions are significantly different between the two samples (P<0.05). Overall, the relative abundance of these functions such as cell wall/membrane/envelope biogenesis, nucleotide transport and metabolism, coenzyme transport and metabolism, translation, ribosomal structure and biogenesis, replication, recombination and repair, defense mechanisms, etc. in the BM1 sample is significantly higher than the M1 sample (P<0.05). On the contrary, the relative abundance of lipid transport and metabolism, transcription, energy production and conversion, amino acid transport and metabolism, organic ion transport and metabolism in the M1 group was significantly higher than the BM1 sample (P<0.05) (Figure 7).

Figure 7: COG function classification statistics chart. Note: Different colors in the picture represent different groups. The left figure in the picture shows the abundance ratio of different functions in two samples or two groups of samples, the middle shows the difference ratio of the function abundance within the 95% confidence interval, and the rightmost value is the p value.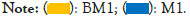
Microbial communities associated with plants can have positive and negative effects on plant health and nutrition as well as beneficial to the global carbon and nitrogen cycle [16]. Soil microorganism’s communities also play an important role in crop health and disease. Therefore, understanding and studying the composition and changes of mango rhizosphere soil microorganisms is helpful to analyze the impact on mango healthy cultivation. High-throughput sequencing technology has been widely used in the research of rhizosphere soil microorganisms. Compared with traditional molecular biology methods, high-throughput sequencing technology can more comprehensively reveal relevant information about the structure of soil microbial communities. This study used high-throughput sequencing technology to conduct microbial amplicon sequencing analysis. Through comparative analysis, we found detailed changes in soil microbial communities between diseased and normal mango plants, and revealed how these changes affect the growth and development of mangoes.
Rhizosphere microbial community
The rhizosphere of plants is the most direct and intense area of the root systems own life activity and metabolism to the soil. Bacteria, fungi and other microorganisms in soil are attracted by plant root exudates and enriched in rhizosphere soil, which plays an important role in plant growth and development. Other studies revealed that the microbiota associated with plants is crucial in determining plant performance and health, because certain beneficial microbes improve plant growth and resistance to stresses.The microbial communities in the rhizosphere soil of plants are dominated by more rapidly growing nutrient-rich bacteria, such as Proteobacteria, Bacteriodetes, Firmicutes, Actinobacteria [17], which can release and help roots absorb available potassium, phosphorus and other micronutrient elements plants [18].
This experiment shows that the microbial community structure and diversity in soil samples from diseased mangoes and normal mangoes collected in the same field are similar but different. The two soil bacterial groups collected in Baise are mainly concentrated in Proteobacteria, Acidobacteria, Verrucomicrobia, Bacteroidetes. Despite the large number of bacterial phyla described in nature and the multiple factors that affect these communities, the bacterial microbiota of plants is dominated by three major phyla (Proteobacteria, Actinobacteria, and Bacteroidetes) in both above and belowground plant tissues. Compared with normal samples, the abundance of Proteobacteria and Verrucomicrobia in diseased samples decreased, and the abundance of Acidobacteria and Bacteroidetes increased. This may be related to mango root exudates. There are many types of plant root exudates, and root exudates not only provide energy for the rhizosphere soil microbial system, but also affect the diversity and richness of rhizosphere microorganisms [19]. Plant-specific root exudates display the specific selection of rhizospheric microbial communities [20]; for instance, cucumber plant secreted citric acids from its roots, which then influenced the attraction of Bacillus amyloliquefaciens and banana root exuded fumaric acid, which attracted B. Bacillus subtilis toward roots leading to biofilm formation [21]. At the same time, studies have shown that there are some substances in the root exudates that inhibit the growth of soil microorganisms, such as phenolic compounds, hydrocyanic acid, ferulic acid, P-hydroxybenzoic acid, etc. [22]. Through the analysis of KEEG metabolic pathways, this study found that the BM1 sample microbial community has a higher relative abundance in the metabolic pathways such as biosynthesis of other secondary metabolites, glycan biosynthesis and metabolism, metabolism of cofactors and vitamins, and energy metabolism. The COG function prediction analysis also showed that the BM1 sample sequence was found to have higher relative abundance in coenzyme transport and metabolism, signal transduction mechanisms, carbohydrate transport and metabolism and other functions. Therefore, it is possible that the plant's disease caused the plant to secrete a certain substance that inhibited the growth of the microbial community, resulting in a change in the diversity and richness of the community of normal samples and diseased samples. This is consistent with the previously reported results that the microbial community in the soil is affected by plant root exudates and plays an important role in the growth and development of plants.
Microbial interaction
Among the soil microorganisms, Sphingomonas has been reported to have a certain growth-promoting effect [23]. In the diseased samples, the abundance of Sphingomonas was significantly less than that of the normal samples. Obviously, under the same conditions, the microbial community with growth-promoting function decreases, which leads to the decrease of the activity of beneficial microorganisms in the soil, which may be the cause of mango disease. Compared with non-rhizosphere soil, the soluble root exudates of plants provide abundant available carbon sources for rhizosphere microorganisms, and the chemotactic reaction of some bacteria, such as Proteobacteria, Bacteroides and Actinobacteria, to carbon sources is the first step of their colonization in roots [24,25]. By studying the utilization of root exudates by strains isolated from the rhizosphere of plants, it was found that these rhizosphere-isolated strains mainly use amino acids, carbohydrates and nucleotides. Sugars and amino acids in the rhizosphere of plants are easily assimilated by microorganisms, forming an environment with abundant carbon sources, Stimulating the chemotaxis of Plant Growth Promoting Rhizobacteria (PGPR) of root surface plants such as Bacteroides and Proteobacteria, and affecting the recombination of soil microbial populations in the rhizosphere in terms of type and quantity. In this study, the abundance of Bacteroidetes and Proteobacteria in diseased samples was lower than that of normal samples. This may be due to the obstruction of root secretion of diseased plants, which affected the trend of rhizosphere soil microbial communities, resulting in lower abundance of growth-promoting microbial flora.
Effects of soil physical and chemical properties on rhizosphere microorganisms
The growth and water absorption capacity of plant roots directly affect the physical properties of rhizosphere soil. The pressure generated by the growth of plant roots will change the rhizosphere soil porosity, soil strength and other physical properties, thereby affecting the rhizosphere microbial community composition. Concentrating on severely phosphorous drained soils [26] showed that the extent of non-mycorrhizal plant species expanded directly with phosphorous deprivation in soils. Field experiments have shown that the increase of soil strength increases the number of wheat rhizosphere bacteria, and the change of soil structure is also an important factor affecting Rhizoctonia solani [27].
Soil pH is one of the main reasons for changing bacterial diversity and community composition. The deviation of environmental pH from neutral is not conducive to the survival of microorganisms, which may be related to the narrow adaptation range of microorganisms to soil pH. Soil acidification will reduce the microbial biomass and microbial activity. If the pH value of chickpea rhizosphere soil drops from 5 to 4.3, the microbial activity is inhibited [28]. Root exudates dicarboxylic acid enhances the availability of orthophosphate and micronutrients in the soil by acidifying the soil, leading to an increase in the relative abundance of β -proteobacteria and actinomycetes. In addition, plants can also influence the rhizosphere microbial community by regulating gene transcription and releasing specific root exudates. For example, various hydrolytic enzymes in plant root exudates play an important role in the cycle of carbon, nitrogen, phosphorus, sulfur and other elements, and affect the composition of the rhizosphere microbial community by changing the soil nutrient concentration [29].
The research on the diversity of mango root microorganisms is the basis for the development of microbial inoculants, bacterial fertilizers and the application of biological control of diseases and insect pests in the agricultural field. This article describes the changes and analysis of the diversity of diseased soil and normal soil microbial communities in mango fields in Baise, Guangxi, and provides a theoretical basis for the prevention and control of mango plant root diseases in this area. The next step will conduct an in-depth comparative study of the mango rhizosphere microbial diversity in Baise mango and even the main mango growing areas in China (Hainan, Sichuan, Yunnan and other provinces and its relationship with the physical and chemical properties of the soil and nutrient elements. It is also our plan to develop beneficial microbial resources that can be used for mango plant cultivation and planting to improve the resistance, yield and quality of mangoes.
This work was supported by the Science and Technology Project of Guangxi (2020ZYZX3027, AB2107601), and a Bagui Scholar Program Fund (2016A25) of Guangxi Zhuang Autonomous Region awarded to LQ.Z. We appreciate all scholars enrolled in this study, including the Bagui Scholarship of Guangxi Zhuang Autonomous Region awarded to LQ.Z.
The authors declare that they have no conflict of interest.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Wang X, He S, Zhu Q, Ye L, Wei S, Zhou L (2022) Comparison Study of Correlation between the Structure of Microbial Communities in Mango Plant Roots. J Prob Health. 10:287.
Received: 01-Aug-2022, Manuscript No. JPH-22-19159; Editor assigned: 03-Aug-2022, Pre QC No. JPH-22-19159 (PQ); Reviewed: 17-Aug-2022, QC No. JPH-22-19159; Revised: 24-Aug-2022, Manuscript No. JPH-22-19159 (R); Published: 02-Sep-2022 , DOI: 10.35248/2329-8901.22.10.287
Copyright: © 2022 Wang X, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.