Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Research Article - (2015) Volume 4, Issue 2
Anthocyanins are a group of secondary metabolite that is responsible for the color in plant and their antioxidant activity has been noticed as a health-promoting material. To identify the genes responsible for anthocyaninenrichment in the black rice grains, we assessed the expression of transcripts in both black and white rice cultivars using the 135K Oryza sativa microarray and found that the 3,728 genes were associated with the production of anthocyanin pigment. Among them, the 573 conserved orthologous genes were identified using the COGs analyses and were compared with the existing flavonoid-pathway network. Enriched-pathway analysis finally resulted in 53 candidates for anthocyanin biosynthesis. These genes were anchored to the chromosomes of the rice genome to identify their genetic-map positions and were subjected to the phylogenetic construction together with their 31 homologous proteins sequences from A. thaliana, using the maximum-likelihood method. Our candidate genes seem to either play a regulatory role in anthocyanin biosynthesis or be related to anthocyanin metabolism.
<Keywords: Anthocyanin; Flavonoid; Phylogenetic tree; Rice microarray
Anthocyanins, a member of flavonoids, are the primary pigments in grains, flowers, fruits, and vegetables, showing blue, purple, and red. It is estimated that there are more than 400 naturally occurring anthocyanins [1]. With their functional impact, their biosynthetic pathway, transportation and regulatory mechanism have been extensively studied in various plant species [2]. The structural genes in their biosynthesis are placed into the general phenylpropanoid pathway, the early flavonoid pathway and the late anthocyanin specific pathway [3]. Genetic, biochemical, molecular and genomic studies have identified the transcriptional regulation of anthocyanin biosynthetic pathway. Recently, members of the anthocyanin family have been recognized as health-promoting food ingredients, due to their antioxidant activity. Furthermore, numerous cell-culture studies have suggested that anthocyanins may function in cancer prevention, obesity control, and health promotion [4]. As a staple food, anthocyanin-enriched (Oryza sativa L.) is one of important goals in the rice breeding program. It was reported that black-colored rice is rich in anthocyanin and its main anthocyanin pigments are composed of yanidin and peonicin [5]. Also, the expression of conserved anthocyanin-biosynthetic genes such as phenylalanine ammonia lyase (PAL), chalcone synthase (CHS), flavonone 3β-hydroxylase (F3H), dihydroflavonol reductase (DFR) and anthocyanin synthase (ANS) were examined in rice [6]. Here, we investigated expression of genes in black rice, in order to gain insight into the mechanism of anthocyanin production. Results of these studies will facilitate future breeding with black rice that is enriched by anthocyanin.
Rice accessions
We utilized microarray data from six rice accessions at three different developmental stages of seed (i.e., 7, 14 and 21 days after heading date). One white rice accession Dongjin and two black rice accessions Heugjinju and Heugseol were acquired from the RDA-Genebank (http://www.genebank.go.kr/) and were cultivated in a standard manner in an experimental field of the National Academy of Agricultural Science (NAAS, http://www.naas.go.kr) [7]. In addition, we analyzed the microarray data from three Ac/Ds gene trapped lines (Ds40112, Ds22978 and Ds97389) expressing high levels of anthocyanins in Dongjin genetic background [8,9].
Microarray data collection
Microarray data were collected from the agricultural crops research project of the NAAS. In total, we investigated gene expression in six accessions (i.e., one white and five black rice cultivars) at three developmental stages based on microarray expression intensity [7,8]. Microarray analysis was performed using the 135K Oryza sativa microarray, which is referenced to the annotation data on the Os-Nipponbare-Reference-IRGSP-1.0 at the Rice Annotation Project (http://rapdb.dna.affrc.go.jp/). In order to minimize the experimental bias resulting from dye swap, gene expression was assessed using only the Cy3 value, and intensity was calculated as the median value of the spot.
To analyze orthologs, Clusters of Orthologous Groups (COGs) analysis were conducted using the eukaryotic NCBI/COGs database (http://www.ncbi.nlm.nih.gov/COG/). We identified the most likely chromosomal positions of each gene using the FSTVAL program [10] and then located the best mapping position in FSTVAL with the BLASTN program, employing an e-value cutoff ≤ 1.0 × 10-5. To identify the interaction pathway networks, we performed an enriched-pathways analysis and Fisher’s exact test to determine the most significant pathway responses using Pathway Studio® software 9.0 (Ariadne Inc., Rockville, MD, USA). Phylogenetic trees were constructed using the maximum likelihood algorithm implemented within the MEGA6.06 program (http://www.megasoftware.net/). The phylogeny of tree nodes was generated with 1,000 bootstrap repetitions, and bootstrap values (above 50%) are given at the nodes; branch values lower than 30% are hidden.
Identification of differentially expressed genes (DEGs) in anthocyanin-accumulating rice accessions
In order to elucidate genes responsible for anthocyanin biosynthesis in rice grains, microarray analysis was previously performed on six rice accessions (i.e., white and black rice) expressing different levels of anthocyanins by using the 135K Oryza sativa array [7,8]. Comparison among three cultivars (Dongjin, Heugjinju and Heugseol) identified 12,673 candidates, while analysis of three Ac/Ds gene-trapped lines and their wild-type Dongjin identified 6,739 genes for flavonoid pathway [7,8]. With an aim to further narrow down to the potential genes in anthocyanin pathway, we integrated these two sets of data and found that 3,728 genes were shown to be either up- or down-regulated with more than 2-fold changes in the five black rice accessions at all three developmental stages, compared to the white rice Dongjin. We observed that the number of up-regulated genes in the black rice was larger than that of down-regulated genes.
Analysis of gene ortholog and KEGG pathway
To examine the function of DEGs in the black rice, we performed Clusters of Orthologous Groups of proteins (COGs) analyses on the 3,728 candidate genes, using the NCBI/COGs database. The up-regulated genes were mainly classified into three categories such as general function (19.7%), signal transduction (15.4%), and chaperone function (10.9%). Genes annotated by COGs analysis were further compared to our unpublished rice database at the NABIC genome center (http://nabic.rda.go.kr/) to eliminate genes that were more related to other pigments (i.e., tannin, phenolics, etc.) from the our gene list. Ultimately, we identified 573 candidate genes that were predicted to be significantly involved in anthocyanin biosynthesis and metabolism. The 573 anthocyanin-related candidates were mapped to the flavonoid-biosynthesis map 00941 of Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/). As expected, genes in the anabolic pathway were up-regulated (Figure 1), whereas those in the catabolic pathway were down-regulated in the black rices (Figure 1, blue text and square). Specifically, the transcript expression of flavanone 4-reductase, dihydrokaempferol 4-reductase, dihydroquercetin reductase, dihydromyricetin reductase and anthocyanidin synthase was increased. However, the expression level of anthocyanin-degrading enzyme, anthocyanidin reductase was decreased, which resulted in the accumulation of anthocyanin and consequently black color in the rice grains.
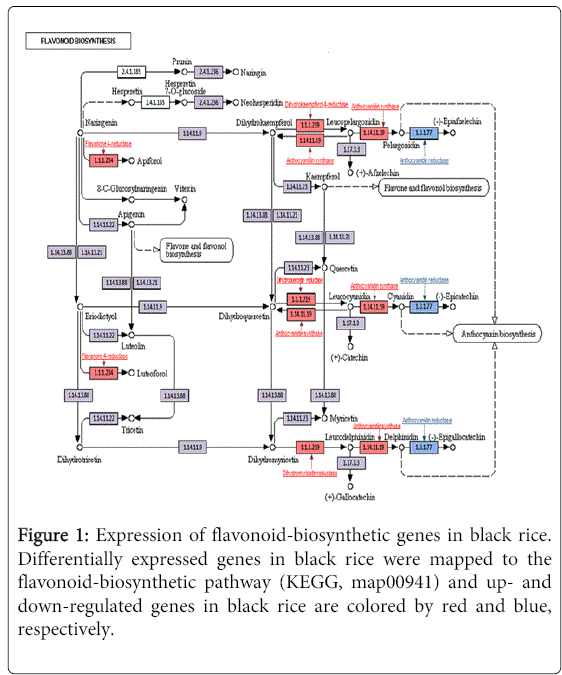
Figure 1: Expression of flavonoid-biosynthetic genes in black rice. Differentially expressed genes in black rice were mapped to the flavonoid-biosynthetic pathway (KEGG, map00941) and up- and down-regulated genes in black rice are colored by red and blue, respectively.
Pathway analysis and genomic localization of anthocyanin-related genes
The 573 DEGs were further subjected to the enriched-pathways analysis by Pathway Studio® software, which finally resulted in 53 genes involved in anthocyanin-related pathways. The positions of 53 DEGs on the 12 chromosomes were identified by mapping the nucleotide sequences of microarray spots to the rice reference genome (Figure 2). The sequences were found to be mapped to one of the three sequence types (i.e., exon, intron, and intergenic), as shown with different colors in Figure 2. Although it is further addressed if the accumulation of intronic and intergenic sequences indicates the increased expression of particular genes, spotting intronic and intergenic sequences on microarray resulted in the detection of protein non-coding regions. Along with the novel genes, our analysis results included the previously reported genes in anthocyanin pathway (Table 1), suggesting that our dataset represents anthocyanin biosynthetic and metabolic pathway. The product of Os01g372500 is a predicted anthocyanidin synthase (ANS) [11], while Os04g0630800 encodes a protein similar to anthocyanidin reductase. In addition, Os02g0289500 and Os04g0557200 encode proteins similar to anthocyanin 5-aromatic acyl transferase and anthocyanin regulatory B protein, respectively. Three other genes encode proteins associated with flavonoid biosynthesis [12,13] (Table 1).
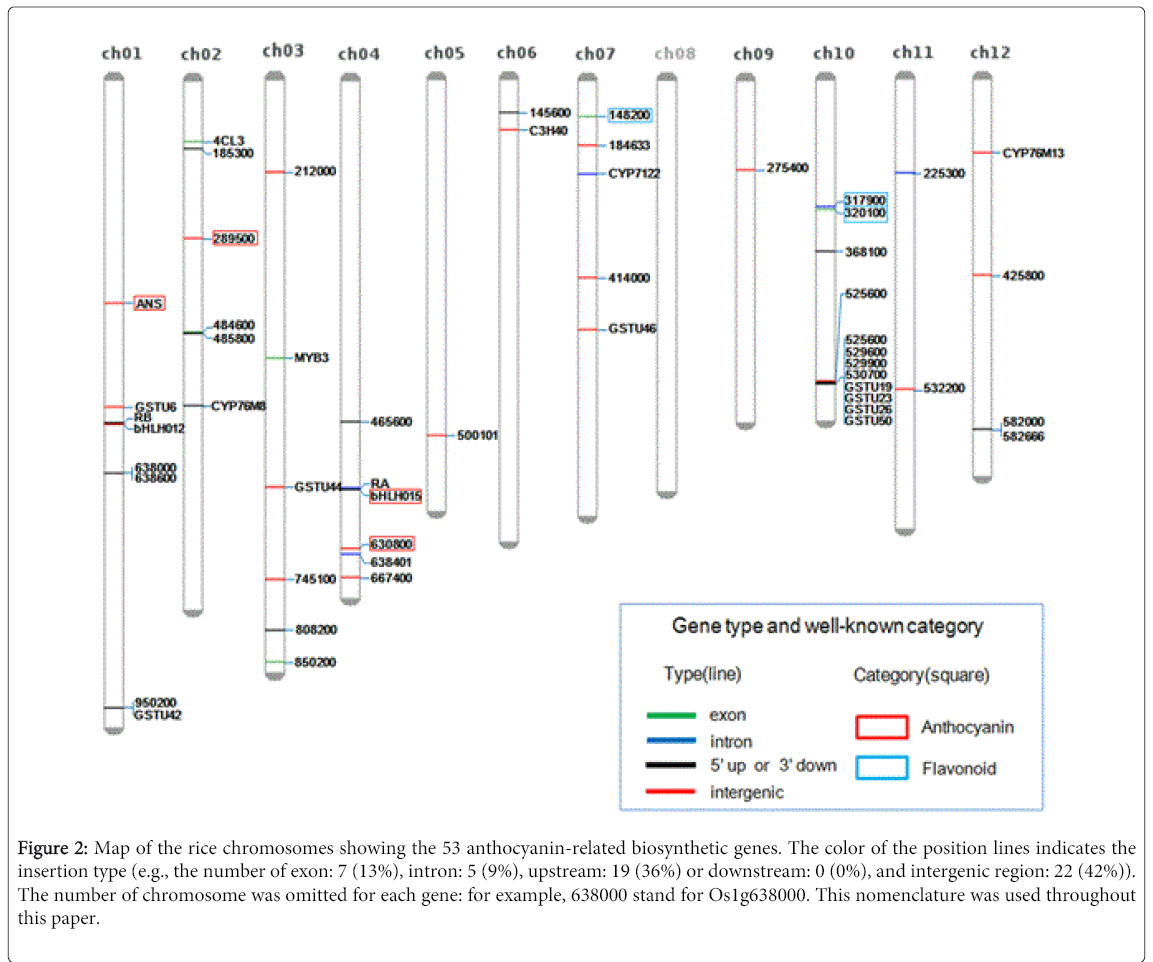
Figure 2: Map of the rice chromosomes showing the 53 anthocyanin-related biosynthetic genes. The color of the position lines indicates the insertion type (e.g., the number of exon: 7 (13%), intron: 5 (9%), upstream: 19 (36%) or downstream: 0 (0%), and intergenic region: 22 (42%)). The number of chromosome was omitted for each gene: for example, 638000 stand for Os1g638000. This nomenclature was used throughout this paper.
| Name | Chra | MLb | Chromosome | Score | RAP-DBc | Type | |
|---|---|---|---|---|---|---|---|
| start | end | ||||||
| ANS | chr01 | 3293 | 15578032 | 15581324 | 6284 | Os01g0372500 | Intergenic |
| 289500 | chr02 | 2163 | 10971914 | 10974076 | 4234 | Os02g0289500 | Intergenic |
| bHLH15 | chr04 | 3590 | 28382617 | 28386206 | 6688 | Os04g0557200 | Intron |
| 630800 | chr04 | 4911 | 32583628 | 32588538 | 9129 | Os04g0630800 | Intergenic |
| 148200 | chr07 | 2905 | 2521346 | 2524250 | 5444 | Os07g0148200 | Exon |
| 317900 | chr10 | 3367 | 8835537 | 8838903 | 6407 | Os10g0317900 | Intron |
| 320100 | chr10 | 2274 | 9016637 | 9018910 | 4419 | Os10g0320100 | Exon |
Table 1: Mapping matrix of the seven well-known anthocyanin-related genes on the rice reference genome. a Chromosome; b Matching length; c Predicted gene name by rice annotation project database (http://rapdb.dna.affrc.go.jp/).
Phylogenetic analysis of anthocyanin-specific genes
To identify the phylogenic relationships among the 53 candidate genes, we performed a maximum likelihood phylogenetic analysis, using MEGA6 software. The resulting phylogenetic tree illustrates that these anthocyanin-related genes cluster into two subgroups (Figure 3). We next wanted to determine the phylogenic relationship among these 53 candidate genes and their homologous genes in A. thaliana, as this comparison with well-characterized homologs might reveal functional insights. To do this, we first identified 31 homologous genes from the A. thaliana genomes using our screened gene sequences. Further phylogenetic reconstruction was then performed with the anthocyanin-related 53 candidate genes and the 31 homologous proteins sequences from A. thaliana, using the maximum-likelihood method. A composite tree indicates the relationships between the genes in rice and Arabidopsis, and all of the anthocyanin-related genes were found to cluster into three subgroups (Groups I to III). Among the 84 selected genes, Group I mainly contains genes up-regulated during anthocyanin biosynthesis, Group II contains genes related to the biosynthesis of other compounds, and Group III contains genes that are both up- and down-regulated during anthocyanin metabolism (Figure 4). The overall clustering pattern of the genes shows a similar tree topology, as observed in the composite tree in Figure 3. Taken together, these results suggest that the screened genes share genetic properties with the previously characterized orthologs involved in anthocyanin biosynthesis-related pathways.

Figure 3: Phylogenetic relationship among the 53 candidate genes predicted to be involved in anthocyanin biosynthesis. Maximumlikelihood phylogenetic trees were generated using the MEGA6 program.
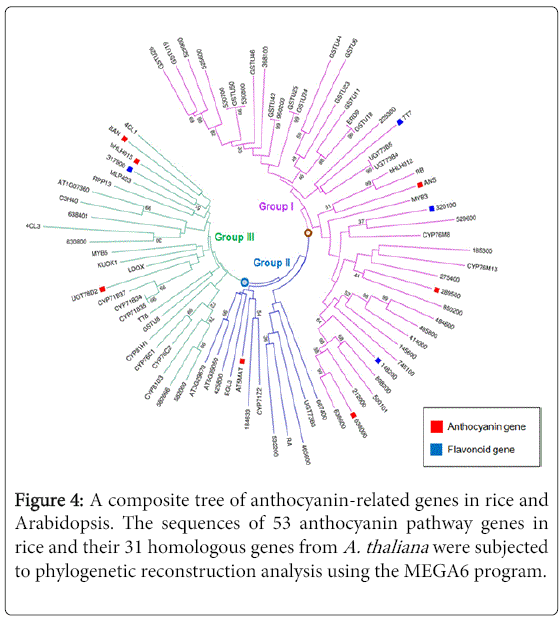
Figure 4: A composite tree of anthocyanin-related genes in rice and Arabidopsis. The sequences of 53 anthocyanin pathway genes in rice and their 31 homologous genes from A. thaliana were subjected to phylogenetic reconstruction analysis using the MEGA6 program.
There has been increased interest in the use of black rice due to the numerous health benefits associated with the anthocyanin pigment. In this study, we identified putative genes involved in anthocyanin biosynthesis in black rice cultivars, using a 135K Oryza sativa microarray. We observed the presence of commonly expressed genes among multiple black rice cultivars and further analyzed them by using currently available database and tools such as COGs, KEGG, ortholog and enriched-pathway analysis, etc. Particularly, we identified 53 genes which may play a regulatory role in anthocyanin production or be related to anthocyanin metabolism during the flavonoid biosynthesis. While these genes require further validation, the results here underline the potential use of the 135K rice microarray and provide valuable insight into the usage of our anthocyanin-rich varieties in the rice breeding program.
This study was conducted with the support from the Research Program for Agricultural Science and Technology Development (Project No. PJ010112) of the National Academy of Agricultural Science, Rural Development Administration, Republic of Korea.