Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2012) Volume 5, Issue 10
This paper investigates epistatic scenarios in a genome-wide association studies (GWAS) context using a qualitative association model, to assess the statistical models that reliably predict associations between a qualitative phenotype (i.e., a disease diagnosis) and a pair of interacting genes. We employed the concept of relative risk, which is the ratio of the probability of a positive diagnosis given a mutated genotype divided by the probability with no risk present.
We used a Monte Carlo-based simulation approach, to generate synthetic data corresponding to a variety of possible epistatic models (EMs). Our method took into account the strength of association, disease prevalence in non-risk populations and most importantly, the inheritance patterns of the epistatic genes. We analyzed the simulated gene data, to assess how these individual factors influenced statistical power in the context of GWAS.
Using simulated data provides two distinct advantages. First, the association-affecting factors are isolated and can be linked to the affecting locus. Second, we can use any specific statistical method to perform the assessment. The simulated dataset provides a truth set, for assessing the effect of statistical method choice on association sensitivity, and highlights the role of errors in disease diagnosis and incorrect genotype assignments.
The results indicate that the most powerful statistical methods for predicting associations between phenotypes and genotypes, in epistatic scenarios are statistical models that simultaneously test for associations involving both interacting loci. This result is not surprising and has been reported by others. Two-gene models produce better predictions of association than single-gene models. The significance of this study is twofold: First, it incorporates recent new statistical methods as part of the comparison analysis and second, it documents the extent to which single-gene models fail to predict associations, involving interacting genes with phenotypes constructed to be associated with low risk.
Keywords: GWAS; Simulation; Epistasis; Case Control; Heritability
The process leading from genes to phenotypes is complex, and the environment can have a large effect. For example, most proteins are the products of multiple genes. Whether a protein is an enzyme, receptor, hormone, or other, it functions in a specific environment that includes external factors (e.g., temperature, rainfall, amount of sunlight, nutrition) as well as internal factors (e.g., other hormones, enzymes). Further, biochemical pathways are not always linear processes: they can have multiple positive and negative feedback loops, may involve multiple steps and the products of hundreds of genes. Thus, the evolutionary forces acting on a single gene are most often not simple, but are environment-specific selection in the context of other gene products.
Recent efforts to unravel the genetic factors that influence important phenotypes such as disease diagnosis and predisposition, have taken the form of genome-wide association studies (GWAS). If genetic variations are more frequent in persons with a given disease, the variations are said to be "associated" with the disease. The associated genetic variations serve as pointers to regions of the human genome, that are potentially involved in causing the disease. The GWAS approach is usually non-hypothesis driven. It uses brute force methods to scan the entire genome, to determine which genes demonstrate an association.
In general, GWAS apply univariate statistical tests to each gene marker or single nucleotide polymorphism (SNP) as an initial step. This SNP-based test is statistically straightforward, and the core tests for assessing the associations are standard methods (e.g., χ2 tests, regression) that have been studied outside of the GWAS context. A recent paper by Kuo and Feingold [1] describes the most commonly used statistical methods that are applied to GWAS. All tests cited in the paper are single-locus tests. Some authors [2] recommend combining two or more statistical tests, if the genetic inheritance properties are not known. In many cases, the SNPs associated with a disease are not in a region of DNA that codes for a protein. Instead, they are in the large non-coding regions between genes or in intron sequences, which are edited out of mRNAs prior to translation to proteins. These regions are presumably, sequences of DNA that modify gene expression, but usually their function is unknown [3].
The popularity of the GWAS approach belies its simplicity and obscures the important issue of, whether a single-gene model is conducive to unraveling the workings of the biosynthetic pathways of a phenotype. In the path leading from gene to trait, factors such as epigenetics, alternate splicing, gene expression levels and proteinfolding processes create a great deal of complexity. These are ignored by qualitative trait analysis, the most common GWAS model reported in the literature. As of mid-2011, over 1,000 human GWAS have examined more than 210 diseases and traits and reported over 1,200 SNP associations. Most of these GWAS employed a single-gene model that assumes that, each locus acts independently of the others.
Many researchers believe that complex diseases involve multiple genes and their interactions [4,5]. Although GWAS have had some success in identifying genetic variants underlying complex diseases, most existing studies are based on limited single-locus approaches, which detect SNPs based on their marginal associations with a qualitative disease diagnosis.
Classical statistical tests derived from case-control experiments involving two loci that use Pearson’s χ2 test or logistic regression, are commonly used as single-locus tests for GWAS and can be used in searching for pair-wise interactions. One early study that investigated interactions is Marchini et al. [6], which showed that explicitly modeling interactions between loci for GWAS, with hundreds of thousands of markers is computationally feasible. They also showed that simple methods explicitly considering interactions, can actually achieve reasonably high power with realistic sample sizes, under different interaction models with some marginal effects, even after adjustments for multiple testing using the Bonferroni correction. However, the genotype-phenotype scenarios addressed by Marchini et al. [6] had substantially larger effects than those that we examine here. Specifically, we focus on low-effect loci, those with low relative risk of association with disease diagnosis, because the evidence suggests they are common [7]. We also focus on theoretical examples of epistasis that are affected by the mode of inheritance, without assuming an additive inheritance model.
An overarching goal of this study was to review the evidence, as to whether statistical methods based on single-gene models can effectively identify genotype-phenotype associations for multi-gene processes. Detecting such associations is particularly difficult for genetic variants with modest impacts on risk. Consequently, our experiments specifically investigated scenarios involving low-risk genetic variants, and assessed whether multi-gene scenarios could be a source of the “missing heritability” observed using single-gene models [8]. We also examined the impact of two recent studies that collaborated in the development of novel tests, for measuring interaction between two linked (in epistasis) or unlinked loci [9,10]. These studies purport to have higher power to detect interaction than classical logistic regression models.
Our investigations demonstrate that for low-effect loci, single-gene models of association fail to identify many associations because the interacting locus masks the effect on the index locus. For the scenarios we tested, our results also support the assessment by Wu et al. [9] and Ueki et al. [10] that, analytical methods that assume statistical interactions between loci are more powerful than single-loci models.
Note that, we will refer to markers as loci but they could also be viewed as genes, SNPs or haplotypes.
Epistasis analysis
One way to extend the single-gene model to accommodate multiple genes, involves studying gene pairs and their epistatic relationships. Epistasis analysis is the genetic methodology used to identify which genes act in a particular cellular process or pathway, and to establish an order-of-function map that reflects the sequence in which they act. It typically involves determining for a pair of genes, whether the phenotype of a double mutant more closely resembles that of one of the single mutants, or if it is a novel phenotype. If a researcher knows what type of pathway is being investigated, this information can help establish what the relationship is between the two genes.
Two types of pathways can be defined: substrate-dependent and switch regulatory. Substrate-dependent pathways consist of a specific series of positive reactions, each of which involves some gene product (e.g., an enzyme) acting on a substrate produced in the previous step in the pathway, and ultimately producing some final outcome. Switch regulatory pathways consist of genes encoding negative or positive regulatory factors that alternate between “on” and “off” states, depending upon upstream signaling events, thereby affecting some downstream response. Because substrate-dependent pathways comprise only positive factors, while switch regulatory pathways can comprise both positive and negative factors, interpreting results from epistatic studies is typically less complex for substrate-dependent pathways. Therefore, for the sake of simplicity, this analysis focuses on substrate-dependent pathways.
A number of studies argue that interacting loci may be normal and not the exception. For example, Templeton et al. [11] report that experience has revealed that, most complex traits depend upon more than one locus [11]. Their study focuses on how often interactions among the loci play a significant role in the mapping from genotype to phenotype, given that the phenotype is influenced by two or more loci. They discuss a number of candidate scenarios, including Coronary Artery Disease (CAD), where the ApoE gene has been shown to affect males and females differently. Even the reported Mendelian trait sicklecell anemia is commonly presented as a single nucleotide trait. Finally, Gilbert-Diamond and Moore indicate that gene-gene interactions (epistasis) are a significant complicating factor in the search for disease susceptibility genes [12].
Objective
This paper investigates epistatic interactions in a GWAS context using a qualitative association model. The purpose of this exercise is to determine the statistical methods and models, that reliably predict associations between a qualitative phenotype (specifically, a disease diagnosis, coded as “case” for a positive diagnosis or “control” for a negative diagnosis) and a pair of interacting genes. As with our other work, we use the concept of relative risk, the ratio of the probability of a positive diagnosis given a specific genotype and epistatic model (EM) divided by the probability with no risk present (i.e., P). The value of P is specified exogenously.
We employed a Monte Carlo-based simulation method to generate synthetic data, corresponding to a variety of possible epistatic models for substrate-dependent pathways. The method takes into account, factors known to influence association measurements in GWAS, including the relative risk of association, disease prevalence in nonrisk populations, inheritance properties of the simulated loci, and most importantly, the epistatic relationship of the simulated loci. We then analyzed the simulated gene data, to assess the influence of these individual factors on statistical power in the context of GWAS. There were two advantages to using simulated data. First, the associationaffecting factors were isolated and could be linked to the affecting locus. Second, we could choose any specific statistical method to perform the association assessment. The simulated dataset provides a truth set for assessing the role of statistical methods on association sensitivity, and highlights the particular role of errors in disease diagnosis and incorrect genotype assignments.
Epistatic models of inheritance
Table 1 defines four possible epistatic models (EMs) for substratedependent pathways, as described in the literature [13]. Let gene1 and gene2 be distinct genes with varying genotypes that affect the production of a common gene product, P, ultimately influencing a phenotype (diagnosis of disease). Mutation of gene1 results in a level of expression X of P and a relative risk Φa of exhibiting the disease phenotype. Similarly, mutation of gene2 results in a level of expression Y of P and a relative risk Φb of exhibiting the disease phenotype. The phenotype of the gene1gene2 double mutant varies according to the EM. If gene1 acts upstream of gene2 in the pathway leading to P (EM 1), the double mutant exhibits the phenotype of the gene1 single mutant (gene1 is epistatic to gene2). Conversely, if gene2 acts upstream of gene1 in the pathway leading to P (EM 2), the double mutant exhibits the phenotype of the gene2 single mutant (gene2 is epistatic to gene1). If gene1 and gene2 function in parallel pathways leading to P (EM 3), the double mutant exhibits a novel, more extreme level of P expression, Z, with associated relative risk Φab. Finally, if gene1 and gene2 act at the same step in the pathway leading to P (EM 4), the observed phenotype can be either one of the phenotypes of the single mutants or a novel phenotype.
| Phenotype of gene1 single mutation |
Phenotype of gene2 single mutation |
Phenotype of gene1 gene2 double mutation |
||
|---|---|---|---|---|
| Model 1 | GENE1 GENE2 → → |
X (Φa) | Y (Φb) | X (Φa) |
| Model 2 | GENE2 GENE1 → → |
X (Φa) | Y (Φb) | Y (Φb) |
| Model 3 | GENE1 → GENE2 → |
X (Φa) | Y (Φb) | Z (Φab) |
| Model 4 | GENE1, GENE2 → |
X (Φa) | Y (Φb) | X (Φa), Y (Φb), or Z (Φab) |
Models 1-4 derived from Anthony Michels, CV (2002) Genetic techniques for biological research: a case study approach. John Wiley and Sons. (http://www.amazon.com/ Genetic-Techniques-Biological-Research-Approach/dp/0471899194)
Table 1: Epistatic Models (Ems) for Substrate-Dependent Pathways.
To simulate the EM scenarios in Table 1, in terms of the contributing locus genotypes, we referred to classical genetics material [14]. In each of the models, there are either two or three possible phenotypes. In our scenarios, there are only two phenotypes (a positive or negative diagnosis), but the risk of a diagnosis depends on the specific pairings of the genotypes. Table 2 outlines the expected risks associated with each possible combination of the wild-type (A and B) and mutant (a and b) alleles of gene1 and gene2 for EM1, taking into account the mode of inheritance (MOI) acting at each locus. Since EM2 is the complement to EM1 and EM3 and EM4 are subsumed by EM1, we limited our analysis to EM1. We used this table to generate synthetic datasets, representing the various scenarios and then examined our ability to link (associate) the phenotypes with the contributing genotypes.
| Epistatic Model | 1 | ||||
|---|---|---|---|---|---|
| MOI | gene1 | D | D | R | R |
| gene2 | D | R | D | R | |
| gene1 | gene2 | Risk (¥) | Risk (¥) | Risk (¥) | Risk (¥) |
| AA | BB | 1 | 1 | 1 | 1 |
| AA | Bb | Φb | 1 | Φb | 1 |
| AA | bB | Φb | 1 | Φb | 1 |
| AA | bb | Φb | Φb | Φb | Φb |
| Aa | BB | Φa | Φa | 1 | 1 |
| Aa | Bb | Φa | Φa | Φv | 1 |
| Aa | bB | Φa | Φa | Φb | 1 |
| Aa | bb | Φa | Φa | Φb | Φb |
| aA | BB | Φa | Φa | 1 | 1 |
| aA | Bb | Φa | Φa | Φb | 1 |
| aA | bB | Φa | Φa | Φb | 1 |
| aA | bb | Φa | Φa | Φb | Φb |
| aa | BB | Φa | Φa | Φa | Φa |
| aa | Bb | Φa | Φa | Φa | Φa |
| aa | bB | Φa | Φa | Φa | Φa |
| aa | bb | Φa | Φa | Φa | Φa |
a = risk allele for gene1
A = allele without risk for gene1
b = risk allele for gene2
B = allele without risk for gene2
Table 2: Epistatic Model 1 depicted in terms of risk associated with various genotype combination.
Generation of synthetic SNP data
The data generation method we employed is applicable only to autosomal genes. Furthermore, because our simulation process assumed epistatic behaviors involving two interacting loci, we expect that the findings would apply to genes exhibiting these types of interactions. We began generating data by considering disease penetrance. We define P as the prevalence of a specific trait due to non-genetic factors. We designate a as the risk allele and A as the allele without risk for gene1. Similarly, we designate b as the risk allele and B as the allele without risk for gene2. Following the procedure of Iles [15], we can then define the risk of disease as the ratio of the probability of a case given a and /or b divided by the probability of a case given no risk allele, which is P.
Ψ = Pr (case /a, b) / P (1)
Generating the synthetic dataset was straightforward, using the relationships between P and risk for the different epistatic categories. Initially, we assigned values to the following variables:
• N = the target number of cases and controls in a given experiment,
• P = the disease prevalence in subjects without genetic risk of a diagnosis,
• Φa,Φb = the relative risks (1.10, 1.15), and
• G = {g1, g2, g3}, a set of genotype distributions obtained from actual SNP data [16].
Our general strategy was to randomly select a genotype and assign a relative risk (Φa,Φb) based on Table 2. Using the prevalence (P) assumption, we then assigned a case or control code (1, 0). A detailed description of the process follows:
1. Using the master genotype distribution G, draw at random, a genotype (g1, g2 or g3) for gene1.
2. Repeat this process for gene2; i.e., draw at random, a genotype (g1, g2 or g3) for gene2.
3. Using Table 2, select the risk value ¥ of a case for the epistatic model being considered.
4. Based on ¥ and P, define the probability of a case to be x = ¥ * P. (2)
5. Using the estimate of x from equation (2), assign a case (0) or control (1) designation at random. Note that, using the twelve different EM/MOI combinations outlined in Table 2 for EMs 1-3, cases should be linked to both genetic loci, and this association should be identifiable via appropriate statistical procedures. Disease risk depends on specific and unknown disease mechanisms. A relative risk of 1.7 is considered strong and is associated with positive replication [17], but a risk of 1.3 is considered to be a realistic assumption for complex diseases [18]. However, many instances of risk < 1.1 are reported in the literature. We limited our focus to a relative risk range of 1.10 to 1.25 and were particularly interested in cases with low relative risk. Note that, implicit in equation (2) is a definition of prevalence as the proportion of cases that are present where no genetic risk is assumed.
6. Continue the process until n1 cases and n2 controls have been generated (note that in this example n1= n2, but the procedure can be tailored to specific n1/n2 targets).
Statistical models
Using the assumptions presented in Table 2, we generated 1,000 replicates of genotypic and phenotypic data for each MOI pair for EM 1, using different sample sizes and risks. We then investigated the power of different statistical models to detect genotype-phenotype associations. We analyzed models that test each gene independently for association with the phenotype, and models that test pairs of genes with and without interaction terms for association.
Single-gene Methods - Cochran-Armitage Trend Test: The Cochran-Armitage (CA) trend test is often used as a genotype-based test for case-control genetic association studies, as described in Purcell et al. [19]. More generally, it is used in categorical data analysis, to detect the presence of an association between a variable with two categories (e.g., a diagnosis) and a variable with k categories (e.g., a genotype). The CA trend test modifies the chi-square test, to incorporate a suspected ordering in the effects of the k categories of the second variable. For example, one could order the number of mutated alleles as “zero,” “one,” and “two” and conjecture that the allele effect will not become smaller as the dose increases.
As described in Zheng and Gastwirth [20], the CA trend test has three flavors. Using the notation in Table 3 below to define the 2x3 table of case-control counts stratified by genotype, a test statistic (T2(x)) for the three variations of the CA trend methods can be defined as:
T2(x) = n [Σ0,1,2 {xi (s ri - r si)}]2 / [r s (Σ0,1,2 n {xixi ni } – {Σ0,1,2 (xi ni)2 })]. (3)
| AA | Aa | aa | Total | |
|---|---|---|---|---|
| Case | r0 | r1 | r2 | R |
| Control | s0 | s1 | s2 | S |
| Total | n0 | n1 | n2 | N |
AA – Major genotype
Aa – Heterozygote genotype
aa – Minor genotype
Table 3: Terms defined in Equations (2).
The variables ri, si and ni in equation (3) are defined in Table 3. The variable xi represents the specific test, namely x0 = 0, x2 = 1 and x1 = .5.
Under the null hypothesis of no association, T2(x) has an asymptomatic χ2 distribution with 1 degree of freedom. We applied the above test to both gene1 and gene2.
Two-gene Models – Pearson Test: The two-gene, case-control test is derived from the classical case-control test of epidemiology described by Jewell [21]. As with all of the tests, this test compares subjects who have a condition (the “cases”) with subjects who do not have the condition, but are otherwise similar (the “controls”). As in the CA test described above, the Pearson Chi Square test is used in categorical data analysis, when testing for the presence of an association between a variable with two categories (e.g., a positive or negative diagnosis) and two variables with k categories (e.g., three genotypes). For this test, the columns are the nine combinations of genotypes and the rows are the two case-control designations. The central idea is to compute the theoretical frequencies for all eighteen cells from the marginal totals, and then test for statistically significant differences between the theoretical and observed frequencies. This test also uses a χ2 test with (nr-1) * (nc-1) = 8 degrees of freedom.
1. Two-gene Models - The method of Wu et al. [9], as refined by Ueki et al. [10]
Wu et al. [9] developed two novel statistics, refined by Ueki et al. [10], designed to test interactions between linked or unlinked loci without including the influence of main effects.
The 2-locus linked test, TIH linked, is defined as:
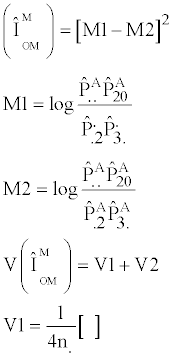
where
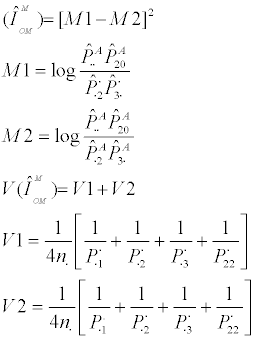
The second test, TIH unlinked, assumes that the two loci are unlinked and is defined as:
where
This study investigated the effect that poly-gene interactions have, on association predictions in a GWAS context. We used statistical models that appear in the literature to generate predictions. Some of the models were single-gene, inheritance-specific models; that is, they assumed that a single additive or recessive or dominant gene produced the diagnosis. Other models were inheritance agnostic and assumed that a pair of interacting genes produced the diagnosis. To implement this investigation, we fixed the risk of the upstream gene of EM1, gene1 to a low but detectable 1.10 risk level. Simultaneously, we varied the risk on the downstream gene, gene2, from 1.00 (no risk) to 1.20, a level that is twice as high as the risk of gene1. Note that, a no risk gene is inconsistent with the purpose of Table 2, which identifies the interactions between two genes. However, we use this scenario to describe an endpoint in our assessment.
Table 4 presents a power analysis for gene1 of the simulated EM1 data, using 6 different statistical tests when the downstream gene has no risk of disease (the single-gene scenario). The first three columns correspond to three different versions of the single-gene Cochran Armitage (CA) test with different inheritance assumptions: additive (CA-A), dominant (CA-D), or recessive (CA-R). Each test was applied to both the upstream and the downstream gene. The last three columns of Table 4 present the results for the two-gene tests.
| MOIs/Stat Model | CA-A gene1 | CA-D gene1 | CA-R gene1 | CC | TIH linked | TIH unlinked |
|---|---|---|---|---|---|---|
| D-D | 59.34 | 73.63 | 0.00 | 50.40 | 0.00 | 0.30 |
| D-R | 59.35 | 73.40 | 0.05 | 51.70 | 0.00 | 0.30 |
| R-D | 9.05 | 0.00 | 23.70 | 9.05 | 0.00 | 0.20 |
| R-R | 7.65 | 0.00 | 23.30 | 9.20 | 0.05 | 27.15 |
Red = Optimal Values
Table 4: Model Comparisons for EM 1, N = 12500, P=.4, Φa = 1.10, Φb = 1.00.
Table 4 indicates the following:
• Single-gene tests work better (from a statistical power perspective) than two-gene tests for single-gene scenarios (i.e., low risk of disease for gene1, no risk for gene2), because the additional degrees of freedom used by the two-gene test, provide no benefit,when there is no additional risk of disease from the second interacting gene.
• In general, the MOI of the upstream gene determines which test is optimal (optimal values are boldface type in red); with the dominant version of the CA test being optimal for dominant genes, and the recessive version of the CA test being optimal for recessive genes. Accordingly, the commonly used additive CA test (CA-A) is never optimal, unless the MOI of the gene is additive [2].
• Unexpectedly, when both genes are recessive, the unlinked refined Wu et al. [9] test is optimal, even though the risk of the second locus is null.
In contrast, Table 5 presents the results for the case, in which the risk from the downstream gene is twice the risk of the upstream gene.
| MOIs/Stat Model | CA-A gene1 | CA-D gene1 | CA-R gene1 | CC | TIH unlinked | TIH linked |
|---|---|---|---|---|---|---|
| D-D | 0.20 | 0.45 | 0.00 | 80.87 | 96.56 | 93.31 |
| D-R | 25.45 | 38.15 | 0.00 | 68.65 | 63.05 | 53.35 |
| R-D | 0.00 | 0.00 | 0.00 | 99.95 | 61.90 | 49.50 |
| R-R | 1.65 | 0.05 | 8.35 | 81.75 | 27.30 | 27.25 |
Red = Optimal Values
Table 5: Model Comparisons for EM 1, N = 12500, P=.4, Φa = 1.10, Φb = 1.20.
The results in Table 5 indicate the following:
• The Pearson two-gene test is optimal for all MOI submodels of the EM1 model, except when both genes are dominant. In this case, the unlinked refined Wu et al. [9] test is optimal; and
• The risk conveyed by gene2 has apparently masked the contribution of gene1 and the power to predict an association between gene1 and diagnosis using single-gene models is very low, below 3% in all cases except submodel (D-R), where it is below 30%. This finding suggests that gene1 is unlikely to be associated with a diagnosis using single-gene models.
Figure 1 provides estimates of the statistical power (Y-axis) to predict the association between gene1 and diagnosis, given different risk values for gene2 (X-axis) for the dominant submodels (D-D and D-R) for EM1. The results presented in Figure 1 correspond to the best single-locus and two-locus tests. Note that, the risk value for gene1 is fixed (1.10). Figure 2 presents the same information for the recessive submodels (R-D and R-R). Both Figure 1 and Figure 2 identify the crossover risk, which is the risk value at which the single-gene (optimal model) and the two-gene model have the same power.

Figure 1: Dominant Gene 1 analysis: D-D Crossover Risk = 1.07, D-R Crossover Risk = 1.12.

Figure 2: Recessive Gene1 analysis: R-D Crossover risk = 1.05, R-R Crossover Risk = 1.07.
These figures suggest that beyond a risk value of 1.05-1.12 (depending on the MOI), single-gene tests are no longer as effective (from a power perspective) as two-gene tests. Furthermore, the power of two-gene tests improves as the risk of the downstream gene increases, whereas the power of single-gene tests progressively declines as the risk from gene2 increases.
We repeated the same analysis for EM 1 with the downstream gene (gene2), risk fixed at 1.1. This time, we varied the risk levels of gene1 and applied the single gene tests to gene2. Table 6 presents the results when the risk from the upstream gene is null (again, we acknowledge that a no risk gene is inconsistent with the purpose of Table 2 but this scenario describes an endpoint).
| MOIs/Stat Model | CA-A gene2 | CA-D gene2 | CA-R gene2 | CC | TIH unlinked | TIH linked |
|---|---|---|---|---|---|---|
| D D | 1.85 | 3.55 | 0.10 | 32.12 | 37.57 | 26.75 |
| D R | 0.10 | 0.00 | 0.65 | 1.15 | 11.10 | 8.45 |
| R D | 41.20 | 56.15 | 0.00 | 52.60 | 11.65 | 8.05 |
| R R | 4.20 | 0.00 | 14.65 | 8.10 | 3.95 | 23.90 |
Red = Optimal Values
Table 6: Model Comparisons for EM 1, N = 12500, P=.4, Φa = 1.00, Φb = 1.10.
Surprisingly, the results indicate that single-gene tests do not universally perform better (in a power sense) than the two-gene tests, even when there is no risk of diagnosis from gene1. Specifically, if gene1 is recessive, the single gene tests do as well as or better than the 8df χ2 two-gene tests, but if gene1 is dominant, the two-gene unlinked refined Wu et al. [9] outperforms the single gene tests.
Table 7 presents the results for the case, in which the risk from the upstream gene is twice the risk of the downstream gene and demonstrates that the two-locus, case-control test outperforms all single-gene tests as well as both of the refined Wu et al. [9] tests in this scenario.
| MOIs/Stat Model | CA-A gene2 | CA-D gene2 | CA-R gene2 | CC | TIH unlinked | TIH linked |
|---|---|---|---|---|---|---|
| D D | 2.00 | 4.40 | 0.10 | 95.36 | 38.22 | 26.70 |
| D R | 5.00 | 0.00 | 0.55 | 100.0 | 11.15 | 8.05 |
| R D | 42.90 | 57.80 | 0.00 | 83.50 | 11.45 | 9.50 |
| R R | 3.70 | 0.05 | 14.00 | 83.80 | 3.60 | 25.90 |
Red = Optimal Values
Table 7: Model Comparisons for EM 1, N = 12500, P=.4, Φa = 1.20, Φb = 1.10.
Figure 3 provides estimates of the power (Y-axis) to detect association between gene2 and disease diagnosis given different risk values for gene1 [1.0 (no risk) to 1.20 (double the risk of gene1)]. Note that the risk of gene2 is fixed at 1.10. Figure 3 presents the results for the dominant gene2 submodels (D-D and R-D). Figure 4 presents the results for the recessive gene2 submodels (D-R and R-R). Figure 3 and 4 are consistent with the results from Figures 1 and 2, and further suggest that beyond risk value = 1.05, single-gene tests are no longer as effective from a power perspective as two-gene tests. Furthermore, the power of two-gene tests improves as the risk of the downstream gene increases. This is exactly the opposite scenario for single gene tests, which decline in power, as the risk of the downstream gene increases.

Figure 3: Dominant gene2 analysis: D-D Cross over Risk = 1.0, R-D Cross over Risk = 1.12.

Figure 4: Recessive gene2 analysis: R-D Cross over Risk = 1.05, R-R Cross over Risk = 1.07.
In this study, we did not vary sample size (N), rather, we fixed N to a high value (12500) to compensate for the fact that we selected low risk loci for investigation. Even for this large value of N, many of the experiments we describe recorded low power values (Table 6). The relationship between sample size and power in a GWAS context has been reported extensively by us elsewhere [22] and demonstrated that, in general GWAS are dramatically underpowered- a significant reason being that both genotype errors and phenotype (diagnosis) errors are assumed to be insignificant (zero).
Our investigation of epistatic scenarios involving low-risk loci indicates that for a given locus, single-locus tests are not as effective as two-locus tests for predicting associations, if the risk value for a second interacting locus exceeds 1.05- 1.12. The cross-over risk value varies, depending on the genetic inheritance properties of the pair of loci. In general, the power of two-locus tests to detect associations improves as the risk value of the second locus increases, whereas the power of single-locus tests progressively declines. Disturbingly, for certain inheritance models and risk values, a true association between a locus and phenotype can be entirely masked by a second interacting locus when using single-locus tests. These findings are not unexpected and are consistent with previous findings reported by Li et al. [4], Culverhouse et al. [23], and Hoh et al. [24], among others. However, single-gene models continue to be used as the core methods for detecting associations in a GWAS context. Our study is significant in that, it provides a more exact estimate of the risk scenarios in which single-locus models are inferior.
Comparing the performance of the three different two-locus tests evaluated in this study, in most cases for EM1, the two-locus, case-control Pearson test is optimal. In certain scenarios (i.e. when both genes have a dominant MOI), the unlinked Wu et al. [9] test (in which cases and controls are included), as refined by Ueki et al. [10] is optimal. This finding is somewhat surprising, given that the modified Wu et al. [9] test measures interaction effects exclusively, whereas the two-locus, case-control test includes main effects for both loci as well as interaction effects.
Despite the widespread recognition that single-locus tests are likely to be inferior to multi-locus tests, for GWAS of many diseases and phenotypes, an unresolved issue is how to construct a computationally practical test that takes into account interactions and enhances the detection of associations between a specific locus and the phenotype of interest. Wang et al. [25] conducted an empirical comparison of five epistatic interaction detection methods, including a number of twopass methods. They indicate that each of the five methods demonstrates unique utilities, but no single method is optimal, being simultaneously the most powerful, the most scalable and having the lowest type-1 error rate in every setting. When users want powerful results and are not concerned with computation cost, Wang et al. [25] cite the TEAM method of Zhang et al. [26] as the best performing algorithm. However, researchers should note that, even when limiting the number of interacting genes to two, n * (n-1)/2 association calculations are required. For n = 500,000-1,000,000, the computational requirements of such an analysis are daunting but readily parallelizable.