Journal of Physical Chemistry & Biophysics
Open Access
ISSN: 2161-0398
ISSN: 2161-0398
Review Article - (2014) Volume 4, Issue 4
Conformations of alpha-substituted pyrroles have been effectively studied using spectroscopic methods assisted by theoretical calculations developed in the recent decade. The question of how to effectively study the conformation of 2-acylpyrrole no longer remains unanswered. The detailed spectroscopic studies conducted in the last decade and interpreted on the basis of theoretical calculations provide a satisfactory answer to that question. Based on the Density Functional Theory (DFT) calculations of conformational properties of 2-acylpyrroles, for which two stable rotameric forms were predicted, syn and anti-conformers have been studied either by experimental or theoretical methods. The family of 2-acylpyrroles have both a proton donor N-H group and a proton acceptor C=O group. This structure favors the formation of doubly hydrogen-bonded cyclic dimers connected by two N-H...O=C bonds. The tendency to form cyclic dimers stabilizes the syn-conformation. Due to these properties 2-acylpyrroles can be used as structural models for the conformational analysis of peptides.
This review summarizes recent investigations of conformations of 2-acylpyrroles, with a particular emphasis on the hydrogen bonds forming within these systems. The influence of 2-substitution on different aspects of stability of these molecular systems and the usefulness of infrared spectroscopy supported by theoretical calculations in H-bonds and conformational studies are discussed. Among the molecular properties hydrogen bond energy, structural characteristics such as C=O bond length of dimers and unique spectral features of 2-acylpyrroles that can be used to predict and investigate the conformation and structure of proteins are considered.
Keywords: 2-acylpyrrole conformers; Hydrogen bonding; DFT calculations; QTAIM; IR spectroscopy; Crystal structure
2-acylpyrrole conformers; syn; Anti conversion; IR; Density functional theory (DFT); Intermolecular hydrogen bonding; Cyclic dimers
Aromatic five-membered heterocycles are an important class of organic compounds. Pyrrole contains a nitrogen which bears a partial positive charge, while the carbon atoms show an increase in electron density as compared with benzene. The most refined description of heterocyclic compounds by Russian physical chemist Nicolay Beketov is noted here, who compared heterocyclic molecules to jewelry rings studded with precious stones. Indeed, the heteroatom gives to a heterocycle its representative properties. The π-excessive pyrrole ring can easily undergo reactions with electrophiles forming predominantly 2-substituted derivatives. Using the Vilsmeier reaction [1] pyrrole yields corresponding pyrrole-2-carboxaldehydes [2] by acetylation pyrrole undergoes selective conversion to pyrrole-2-carboxylic acids [3] and corresponding esters [4]. Pyrrole-2- carboxylates are also useful substrates for the preparation of more complex pyrrole containing systems [5] such as ketorolac, a drug with anti-inflammatory and analgesic properties [6]. It is noted that the 2-acylpyrrole motif is widespread in nature. There are many examples of 2-acylpyrrole based molecules (Figure 1) with various biological activities [7,8]. Representative examples are pyoluteorin and pyrrolomycin D, secondary metabolites produced by marine bacteria [9-11]. There are also certain marine natural products such as nakamuric acid [12], marinopyrroles [13,14] or storniamide which show potent activity as inhibitors of multidrug resistance (MDR) [15] or pentabromopseudodiline [9] which shows good activity against some Staphylococcus aureus strains. Among the bioactive marine compounds there are also distamycin and netropsin [16] with antiviral and antimitotic activities. Due to promising antibacterial properties, these novel marionopyrroles have attracted considerable attention recently [17]. The molecular basis of marinopyrrole biosynthesis that includes a novel N,C-bipyrrole chiral homocoupling reaction has been characterized through a series of genetic experiments. It has been discovered that the chiral coupling reaction of two dichloropyrrole molecules is catalyzed by FADH2-dependent halogenases, enzymes that catalyze electron transfer [18]. Exploration of the mechanistic details of this reaction opens up opportunities for new synthetic routes to bioactive marinopyrroles.

Figure 1: Some of the 2-acylpyrroles from bioactive natural products.
Spectroscopic, theoretical and X-ray studies of 2-acylpyrroles are valuable sources of well documented structural characteristics of a small building block which makes up bioactive natural products. Such studies are important due to the fact that the determination of the structure of large molecules of natural origin is frequently difficult. An example is benzosceptrin [19,20] isolated from Agelas sp, which was synthesized to confirm its putative molecular structure. However, the spectrum of the synthetic compound differs slightly from the spectra of the compound of natural origin [21]. In such cases, it is helpful to know the structure of small structural elements that build large molecules of natural origin [22,23].
It is noted that pyrrol-2-yl carbonyl compounds consist of parallel aligned N-H and C=O groups separated by one carbon atom. It promotes the formation of centrosymmetric dimers with two equivalent N-H…O hydrogen bonds [24]. The dimer-binding motif is strikingly similar to that of the peptide β-sheet. There is a repeat sequence of N-H…O=C hydrogen bonds that help to hold together the sheet-like arrangement of peptide chains. It is reasonable to believe that pyrrol-2- yl carbonyl molecules and its dimers may serve as structural models to study conformations and hydrogen bond patterns in peptides, proteins [25] and other pyrrole-based molecules, such as chlorophyll, hem, plasmodial pigment fuligorubin [26], alkaloid roseophilin [27], which demonstrate the importance for living systems.
In the beginning of the past decade spectroscopic and theoretical research was undertaken to get an insight into the tree-dimensional structure of 2-acylpyrrole. Such knowledge ensures better understanding of the impact of the spatial arrangement of 2-acylpyrrole for molecular properties such as vibrational spectra and hydrogen bond formation. A study on the spectroscopic properties of pyrrole-2-carbaldehyde and methyl pyrrole-2-carboxylate initiated by Chadwick [28,29] and Meakins [30] inspired Grabowski and Dubis group to extend the study to other 2-acylpyrroles.
The scientific research was based on vibrational spectroscopy, X-ray diffraction, and quantum chemical calculations (ab initio and density functional theory) and Bader theory of Atoms in Molecules, QTAIM. A discussion of the energetic and spectroscopic features of stable 2-acyl conformers will be added to the description of their molecular structures.
Quantum chemical calculations
Theoretical calculations have developed into an important tool in conformational research and can greatly support experimental work with quite good accuracy. Among the most useful parameters are geometrical parameters, relative conformational energies, barriers to internal rotation and vibrational frequencies for each conformer.
Energy difference between rotational isomers: Early experimental studies on the conformational preferences for some 2-substituted pyrroles, carried out in the 1970s [30-35] were based on NMR and IR measurements and dipole moment methods. These studies revealed predominance of the syn over the anti-conformer. Using spin-spin coupling constants with H(3) and syn-aldehyde protons, conformer percentages were calculated [32] resulting in 95% of the syn-form. Subsequent NMR measurements of 2-formyl- and 1-alkyl-2-formylpyrrole indicated the percentage share of the synforms amounting to 85 and 60, respectively [29]. The preference of 2-formyl- and 2-acetylpyrrole for the syn conformation in solutions was also confirmed by later studies of the dipole moment [35]. Kaye et al. established, using a variable temperature experiment, that the synform of t-butyl pyrrole-2-carboxylate is thermo chemically more stable by 1.15 kcal/mol than the anti-form [30]. The effects of solvent such as benzene and dioxane on the dipole moments of planar conformers of 2-formyl- and 2-acetylpyrrole were investigated by Lumbroso [36] and Cumper [37] to establish the preferred conformation.
Low-temperature 13C NMR spectra were applied by Lunazzi [38] in the conformational study of thiophenecarbaldehydes. The activation energy for the conversion of the more stabz syn-form into the less stable anti-conformer is 10.1 kcal/mol for thiophene-2-carbaldehyde. Jaureguiberry showed that the free energy of activation for the syn/anti conversion of N-methylpyrrole-2-carbaldehyde is 11.2 kcal/mol [39].
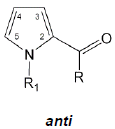
Recent theoretical and IR studies have shown that there are two stable rotational isomers in the liquid state while only one, the energetically most favorable rotational isomer of all those compounds, is found in the solid state [40]. The more stable rotamers of pyrrole-2-yl carbonyl moieties are presented in Figure 2.

Figure 2: (a) Schematic representation of more stable pyrrol-2-yl carbonyl conformers syn and anti where R= H, OH, OCH3, NH2, NHCH3, N(CH3)2, CH2Cl, CHCl2, CCl3; (b) conformation of different types of 2-acylpyrrole dimers, where R= OH, NH2, NHCH3. The dimer represents combination of a 2-acylpyrrole pair which was found in many aggregates presented in Table 2; small circles correspond to ring critical points (RCP).
The syn or s-cis form denotes a rotational isomer in which the carbonyl group is located at the same side of the single C-C bond as the N-H (or N-CH3) groups, anti denotes the carbonyl group being s-trans with respect to the N-H or N-CH3 group. In other words the syn and anti- conformers differ in the spatial arrangement of the carbonyl group about the inverting single C-C bond. Hence, two conformers have to be considered for each compound to determine the possible conformational equilibrium. It is worth noting that theoretical calculations and crystal structure data show that the carbonyl group is, in general, in the same plane as the pyrrole ring. The flat arrangement of the carbonyl group in respect to the pyrrole ring favors the formation of centrosymmetric dimers with two equivalent N-H…O=C hydrogen bonds (Figure 2). In Table 1 rotational isomers of compounds 2-19 and 21-38 discussed in this review are presented. Most of the collected data were Published Date: previously by Dubis and Grabowski [24,40]. Density functional theory calculations of energetic and geometrical properties of molecular structures of the pyrrole-2-yl carbonyl species. Table 1 generally revealed that the syn-rotamers are energetically more favorable than the anti ones [41]. It is in agreement with IR data showing that the concentration of the syn form is higher than that of the anti form [42]. The analysis of data from Table 1 provides some findings about 2-acylpyrrole conformers expressed by ΔEcis/trans=Ecis-Etrans energy differences. ΔEcis/trans of 2-substituted pyrroles vary in a range of 1.06-8.04 kcal/mol. ΔEcis/trans of the N-H moieties (2-19) range from 1.06 to 5.15, whereas those of the N-CH3 moieties [21-38] range from 1.93 to 8.08 kcal/mol. The lowest energy difference ΔEcis/trans of 1.06 kcal/mol was observed for ester (6), whereas the highest energy difference ΔEcis/trans of 8.04 kcal/mol was observed for N-methyl pyrrol-2-yl-chloromethyl ketone 37. It is noted that the ΔEcis/trans of N-H pyrrole derivatives are generally larger than those of N-methyl pyrrole moieties.
| Compound |  |
 |
Selected experimental and calculated IR frequencies [cm-1] | ΔEs-cis/s-trans [kcal/mol] | Barrier height Esyn→anti[kcal/mol] |
Barrier height Esyn→anti [kcal/mol] |
HOMA [au] | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp. | Calc.* | Exp. | Calc. | Exp. | Calc. | |||||||
| nN-H | nN-H | nC=O | nC=O | nC=C | nC=C | |||||||
| 1 | pyrole | 3485 | 3600 | 1532b | 1540 | 0.854 | ||||||
| 2 | R1=H, R=H | 3460 | 3565 | 1659d | 1685 | 1552b | 1553 | 3.84 | 16.21 | 10.38 | 0.923 | |
| 3 | R1=H, R=H | 3576 | 1712 | 1554 | 0.911 | |||||||
| 4 | R1=H, R=OH | 3465 | 3570 | 1671d | 1723 | 1555c | 1556 | 1.11 | 11.3 (13.03) |
11.94 | 0.921 | |
| 5 | R1=H, R=OH | 3587 | 1754 | 1561 | 0.916 | |||||||
| 6 | R1=H, R=OCH3 | 3465b 3480b |
3571 | 1719d | 1705 | 1554b 1557c |
1555 | 1.06 (1.10) | 12.35 (12.43) |
11.36 | 0.917 | |
| 7 | R1=H, R=OCH3 | 3589 | 1701d | 1736 | 1559 | 0.913 | ||||||
| 8 | R1=H, R=NH2 | 3415 3336c |
3559 | 1644c 1603c |
1683 | 1552c | 1550 | 4.91 | 10.17 | 6.60 | 0.917 | |
| 9 | R1=H, R=NH2 | 3582 | 1710 | 1560 | 0.882 | |||||||
| 10 | R1=H, R=NHCH3 | 3571 | 1665 | 1558 | 6.02 | 8.75 | 3.33 | 0.914 | ||||
| 11 | R1=H, R=NHCH3 | 3581 | 1690 | 1548 | 0.875 | |||||||
| 12 | R1=H, R=N(CH3)2 | 3450 | 3552 | 1612 | 1623 | 1548a 1547b |
1544 | 4.95 | 7.03 | 2.07 | 0.911 | |
| 13 | R1=H, R2=N(CH3)2 | 3587 | 1697 | 1560 | 0.886 | |||||||
| 14 | R1=H, R=CH2Cl | 3458d | 3562 | 1662d | 1695 | 1546a 1545c | 1546 | 5.15( 3.1) | 13.9 | 10.8 | 0.923 | |
| 15 | R1=H, R=CH2Cl | 3594 | 1683d | 1714 | 1552 | 0.899 | ||||||
| 16 | R1=H, R=CHCl2 | 3457d | 3561 | 1658d | 1692 | 1543a 1542c |
1546 | 4.19 (2.4) | 13.89 | 11.45 | 0.927 | |
| 17 | R1=H, R=CHCl2 | 3562 | 1683d | 1710 | 1548 | 0.921 | ||||||
| 18 | R1=H, R=CCl3 | 3456d | 3563 | 1670b | 1687 | 1538a 1539b 1538c |
1542 | 2.55 (2.51) | 12.85 | 10,33 | 0.926 | |
| 19 | R1=H, R=CCl3 | 3583 | 1679d 1681b |
1708 | 1545 | 0.922 | ||||||
| 20 | methylpyrole | 1517 | 0.865 | |||||||||
| 21 | R1=CH3, R=H | 1670 | 1689 | 1528b | 1531 | 4.52 (3.47) | 15.31 (15.26) | 11.79 | 0.914 | |||
| 22 | R1= CH3, R=H | 1694 | 1533 | 0.906 | ||||||||
| 23 | R1= CH3, R2=OH | 1670 | 1724 | 1530d 1536b |
1531 | 1.93 | 11.19 | 9.25 | 0.913 | |||
| 24 | R1= CH3, R=OH | 1734 | 1536 | 0.904 | ||||||||
| 25 | R1= CH3, R=OCH3 | 1711 | 1706 | 1532b | 1531 | 1.93 | 10.37 | 8.44 | 0.910 | |||
| 26 | R1= CH3, R=OCH3 | 1717 | 1537 | 0.902 | ||||||||
| 27 | R1= CH3, R=NH2 | 1639c | 1683 | 1529c | 1528 | 5.54 | 9.67 | 7.21 | 0.908 | |||
| 28 | R1= CH3, R=NH2 | 1695 | 1537 | 0.882 | ||||||||
| 29 | R1= CH3, R=NHCH3 | 1667 | 1537 | 4.32 | 8.98 | 4.65 | 0.908 | |||||
| 30 | R1= CH3, R=NHCH3 | 1677 | 1542 | 0.877 | ||||||||
| 31 | R1= CH3, R=N(CH3)2 | 1630d | 1635 | 1537a 1536b |
1529 | 3.85 | 13.51 | 0.902 | ||||
| 32 | R1= CH3, R=N(CH3)2 | 1660 | 1542 | 0.870 | ||||||||
| 33 | R1= CH3, R=CH2Cl | 1662d | 1694 | 1527a 1522c |
1526 | 6.61 (5.68) | 12.74 | 6.67 | 0.909 | |||
| 34 | R1= CH3, R=CH2Cl | 1683d | 1693 | 1527 | 0.886 | |||||||
| 35 | R1= CH3, R=CHCl2 | 1658d | 1692 | 1526a 1524c |
1526 | 6.64 (6.97) | 13.5 | 6.59 | 0.905 | |||
| 36 | R1= CH3, R=CHCl2 | 1683d | 1690 | 1527 | 0.889 | |||||||
| 37 | R1= CH3, R=CCl3 | 1690 | 1524a 1522c |
1526 | 8.08 (8.04) | 11.62 | 4.48 | 0.899 | ||||
| 38 | R1= CH3, R=CCl3 | 1681d | 1692 | 1522 | 0.878 | |||||||
Table 1: Experimental and calculated (B3LYP/6-311++(d,p)) IR stretching frequencies of the C=O, C=C and N-H groups, for syn and anti pyrrol-2-yl carbonyl conformers (1-38). ΔE is the difference between the energies of the syn and anti-form. HOMA [49] is the aromaticity indices.
The rotational isomerism of a five-membered heterocycle containing a carbonyl group was studied theoretically as early as in the 1970s. John et al. showed [43] that for pyrrole-2-carbaldehyde the planar syn-conformation is much more stable than the anti structure. Using ab initio calculations at the LCAO SCF/STO-3G level of theory, they showed that ΔEsyn/anti was 1.86 kcal/mol and that those two conformers were separated by a large energy barrier of 9.36 kcal/ mol. The author suggested that the ΔEsyn/anti value indicated very strong conjugative interactions between the pyrrole ring and the carbaldehyde group.
The conformational properties of diheteroaryl ketones of the furan and thiophene series have also been studied both experimentally and with a theoretical ab initio approach by Benassi [44]. They revealed that solid state structures were very similar to those calculated for conformers with the lowest energy and the calculated conformer populations were in qualitative agreement with measurements in solution.
Itoh [45] showed coexistence of anti and syn rotamers for 2-thiophenecarboxaldehyde through matrix-isolation infrared spectroscopy combined with UV photoexcitation as well as DFT and Coupled Cluster calculations. It was revealed that the syn rotamer was more stable than the anti rotamer for 2-thiophenecarboxaldehydes. The situation is quite different from that of analogous 2-furancarboxaldehyde, for which the anti rotamer is more stable than the syn-one. Based on data obtained through matrix-isolation infrared spectroscopy and calculations at the CCSD(T)/aug-cc-pVTZ level, it was showed that the syn rotamer of 2-thiophenecarboxaldehyde was more stable than the anti rotamer by 1.07 kcal/mol
Internal rotation about the C-C bond: According to the IUPAC terminology rotamers are a set of conformers forming by restricted rotation about a single σ bond [46]. The rotational barrier is the activation energy required to convert one rotamer to another. In the rotation of groups about a bond, the potential energy barrier between two adjacent minima of the molecular entity is a function of the torsion angle.
Early experiments on pyrroles focused mainly on the relative conformer ratio. Syn-Forms prevailed in solution and the solid state. Theoretical studies reproduce well the trend. The gas phase energy difference is in a range of 1.06 – 8.08 kcal/mol at the B3LYP/6- 311++G(d,p) level of theory. Measurements of the barrier in 2-acypyrroles are rare. To date barriers are known in a few solvents and in the gas phase. It should be noted that the rotational barrier is the activation energy required to convert one rotamer to another. The NMR-evaluated free energy of activation for rotation about the C(O)–N bond in 2-N,N-dimethylcarboxamides (12,13) of pyrrole (R=NMe2) was obtained using Eyring plots [47]. The C-N rotational barrier is 14.4 kcal/mol. Davis [47] suggested that the barrier to rotation about the bond arises from resonance interactions between the lone pair of electrons on the nitrogen atom and the electronegative oxygen of the carbonyl group.
Energy barriers separating the syn- and anti-form of 1-alkylated 2-formyl pyrroles have been estimated from [1] H NMR coalescence temperature and 13C NMR [39]. The percentage population of the syn forms of 2-formylpyrrole (2,3) and 2-formyl-1-methylpyrrole (21,22) at room temperature has been estimated as 100 and 90, respectively. The energy barrier of 2-formyl-1-methylpyrrole (21,22) is 11.8 kcal/ mol on the basis of the stereospecificity of long-range coupling constants between the formyl hydrogen and ring hydrogen atoms at positions 4 and 5.
The study of Chieh et al. [48] on the internal rotational barriers of the nitro group for
N-substituted nitropyrroles revealed that the calculated (DFT method at B3LYP/6-311G**) barrier height of N-methyl-2-nitropyrrole is 9.88 kcal/mol, whereas for 2-nitropyrrole it is 13.02 kcal/mol.
Theoretical studies of the torsional potential of 2,2’-bipyrrole [49] revealed that the potential-energy surface of 2,2’-bipyrrole in a wide torsional potential range is rather flat, with the energy difference among critical points of 3 kcal/mol. These data suggest a high degree of conformational flexibility within 2,2’-bipyrrole. B3LYP calculations predict essentially free rotation around the C-C single bond in the 180- 50° range.
Here, the potential energy curve obtained for 2-acylpyrroles as a function of torsional angle is presented. The torsional curves computed at B3LYP levels of theory are shown in Figure 3 while corresponding energies are reported in Table 1.

Figure 3: Relationship between the energy of pyrrol-2-yl carbaldehyde (2) and rotation angle ϕ (pyrrole ring – carbonyl group). The most stable syn and anti conformers differ in the spatial arrangement of the carbonyl group about the inverting single bond (C-C). The calculations were performed at the B3LYP/6- 311++G(d,p) level of theory. 360° scanning of the dihedral angle N-C-C=O was performed with an increment of 5°.
The angle of rotation is zero for the syn conformation; for the angle of rotation of 180° there is the anti conformer. Table 1 presents energies for the syn-conformers and for the highest energies obtained during the rotation of the carbonyl group. The highest energies correspond to angles of rotation of 90° and 270° and may be considered transition states for transformation reactions of conformations from the syn to the anti-form. The barrier heights for those reactions vary from 7.03 to 16.21 kcal/mol depending on the R substituent group. On the basis of previous studies [50,51], we noted that the electrondonating substituent slightly destabilizes the ring system, affecting the aromaticity of the pyrrole ring. The introduction of the -NH2 group as R leads to substantial double bond localization in the pyrrole ring and in consequence to a decrease in its aromaticity. In the case of a chloro substituent (R=14-19 and 33-38), an increase in aromaticity of the pyrrole ring was observed [51]. It is in good agreement with the barrier height (Table 1). The more aromatic compound, the higher the rotational barrier.
For all the 2-acylpyrroles studied here, the rotational profile obtained from theoretical calculations has a similar shape. The profiles show the existence of two energetically most stable syn and anticonformers corresponding to a value of ϕ close to 0 and 180°.
The process of transformation of the syn-form into the anti-form has also been investigated to compare the activation energy of these transitions (Table 1). Figure 3 shows the relationship between the energy of the pyrrole-2-yl carbonyl compound and the angle between the planes of the pyrrole ring and the carbonyl group. The more stable syn-conformation is assumed here as the reference (Figure 2). All the pyrrol-2-yl carbonyl compounds under investigation have in principle two barriers: syn→anti and anti→syn energy barriers.
We can see that the barrier height Ea syn→anti of the N-H moieties (2-19) for the transformation of the syn-conformer into the anticonformer varies in a range of 7.03-16.21 kcal/mol, whereas those of the transformation of the anti into the syn-conformer Ea anti→syn range from 2.07 to 11.94 kcal/mol (Table 1). This may be compared with the Ea syn→anti and Ea anti→syn values for N-methyl moieties (21-38), which range from 8.98 to 15.26 kcal/mol and 4.48-11.79 kcal/mol (Table 1). It is seen that the barrier heights for the syn→anti transformation are larger than those for the anti→syn transformation for N-H and for N-CH3 moieties. These data reveal that the syn-form is relatively more stable than its anti-counterpart. This phenomenon is most likely related to more effective π-electron delocalization in the syn-conformation.
Recently, Ducati has studied the conformational equilibria of 2-acetyl pyrrole and N-methyl-2-acetylpyrrole through the theoretical calculation and IR and NMR spectroscopy [50]. It was found that only syn-conformer is present in solution as well as in the solid state. The activation energy ΔEsyn/anti and ΔEanti-syn, calculated at HF/6-31G(d,p) level of theory, amounts to 7.14 and 5.05 kcal/mol, respectively. The NBO analysis showed that the hyperconjugation effect has a role in stabilizing syn rotamer of 2-acylpyrrole. However, this effect make the smaller contribution to the energy barrier for N-methyl-2acetylpyrrole.
In the case of 2-acylpyrroles the carbonyl group is directly coupled with the aromatic ring. The rotation of the C=O group has a role in changing the nature of the C-C bond connecting the carbonyl group with the pyrrole ring. For the more stable syn and anti planar configurations the C-C bond has a more double bond character due to resonance which keeps the carbonyl group in the same plane as the pyrrole ring. The theoretically calculated geometrical parameters of 2-acylpyrroles [51] clearly reveal elongation of the (C2) carboncarbon (C=O) bond for anti compared with syn-conformers. These observations suggest relatively larger stability of the syn-conformer than its anti counterpart. We can state that this phenomenon results from more effective π-electron delocalization. This was very evidently shown by HOMA aromaticity indices [51]. The data point out that there is a trend that HOMAs for syn conformers are higher than those for anti-conformers.
In the 1990s the origin of inversion barrier in ethane, methanol and methylamine was investigated by the Bader group [52]. It was shown that rotation about the C-C bond induces quadrupole polarization of density in the C-C bond, causing it to lengthen. The lengthening leads to a decrease in the magnitude of the attractive interaction of each carbon nucleus, and this is the origin of the barrier.
Our recent DFT calculations of the geometrical parameters of 2-acylpyrrole conformers show that the bond about which rotation occurs elongates. The structural parameters obtained from optimized geometries (B3LYP/6-311++G(d,p)) for the syn and anti-conformers of 2-acylpyrroles were Published Date: in 2013 [51].
From the comparison of the structural parameters obtained for the two conformers, several differences can be noticed. For example, the distance of the bond (C-C) about which rotation occurs for the anti form is longer than that for the syn-form by 0.001-0.005 Å. By contrast, the carbonyl bond (C=O) of the anti conformer is shorter than that of the syn-conformer by 0.001- 0.01 Å
There are good reasons to believe that the energy barrier is mostly due to a loss of coupling of the carbonyl group with the pyrrole ring, i.e. due to electronic effects. In general, the extent of π-conjugation between the carbonyl group and the ring is the main factor which determines the barrier to rotation. An electron donor attached to the carbonyl group reduces this interaction and hence lowers the rotational barrier (Table 1). Detailed investigations of the 2-acylpyrrole family clearly demonstrate that the conformational characteristics of these systems are sensitive to the impact of the R substituent. This especially concerns pyrrole ring aromaticity and the energy profile. These results indicate that the application of new and very reliable theoretical quantum chemical methods provide conformational characteristics of a broad class of heterocyclic compounds.
Experimental determination of 2-acylpyrrole conformers
A large number of physical methods are employed for conformational studies, including spectroscopic and structural X-ray diffraction methods. The majority of conformational studies have been carried out using NMR and vibrational spectroscopy. This is due to the fact that the spectra can be investigated in all states of aggregation such as vapor, liquid and solid. In this section the IR spectra of 2-acypyrroles with conformational equilibria will be discussed.
IR spectroscopy: IR spectrometry has been especially useful in the study of rotameric equilibria and thermodynamic parameters. Chadwick [53-57] has a huge contribution to the study of rotational isomerism in furan, thiophene and pyrrole derivatives bearing carbonyl-containing substituents. During the past years the author of this review has also been involved in the spectroscopic studies of rotational isomerism in carbonyl-containing pyrrole derivatives. Here the IR aspects of this work will be presented in light of previous findings [29-31].
The IR studies of Chadwick [57] of the conformational preferences of heterocyclic 2-carbaldehydes such as furan, thiophene and pyrrole-2- carbaldehydes revealed the presence of a carbonyl doublet in solution. The components of the doublet were assigned as the syn and antiforms. It is noted that several previous studies had suggested that the carbonyl doublet did not reflect the presence of rotational isomers but indicated splitting due to a Fermi resonance effect [30]. To determine the origin of the carbonyl doublet Chadwick [57] suggested carrying out an experiment using solvents with different dielectric constants and investigation of closely related compounds such as deuterated analogs. It was found on the basis of these experiments that the C=O doublet of furan-2-carbaldehyde forms due to Fermi resonance between C=O and nearby interacting absorption. For some other 2-acylfuranes CO doublet bands were assigned as the syn and anti-forms.
Thiophene-2-carbaldehyde was also investigated by several physical methods to define its conformation [54]. Based on IR [55] and 1H NMR studies [56] it was clearly seen that CDCl3 solution contained very predominantly the syn-form of thiophene-2-carbaldehyde. Early spectroscopic studies of pyrrole-2-carbaldehyde and N-methylpyrrole- 2-carbaldehyde revealed a carbonyl doublet in tetrachloride solution. The components of the doublet were determined by 1H NMR to lead to absorption of the syn and anti-forms with a K(syn/anti) value of 22 at 213 K [29]. It means that N-methyl-2-carbaldehyde exists preferentialy in the syn-conformation. The conformational preferences of some trifluoroacetylpyrroles were also investigated by IR and NMR techniques [57,58]. A variable concentration experiment of trifluoroacetylpyrrole in CCl4 solution revealed that the carbonyl doublet was present even in diluted solution. The authors concluded that the second carbonyl band probably forms due to Fermi resonance. However, 19F NMR experiments clearly demonstrated the occurrence of rotational isomers, but it was not known whether the major component had the syn or the anti-arrangement. It was shown in a subsequent NMR study that the syn form was the preferred conformation of N-methylpyrrole-2- carbaldehyde [59].
Other 2-acylpyrroles such as mono ester derivatives (2-methoxycarbonyl-N-alkylpyrroles) have asymmetric carbonyl singlet bands. The band asymmetry phenomenon was also explained using the Fermi resonance effect. Chadwick stated that 2-monoesters of N-alkylpyrroles exist in solution in only one and the most stable syn-form. Literature data show that trifluoro-2-acetylpyrroles, 2-pyrrolealdehyde [31] and methyl-2-pyrrolecarbaldehyde and their N-substituted analogs have strong preferences for the syn-form. It is reasonable to assume that their ester analogs adopt the same arrangement.
In light of these studies, our attempts have been directed to investigate substituent effects on 2-acylpyrrole conformations and their impact on N-H...O=C hydrogen bond formation (Figure 2). This was explored using spectroscopic methods supported by theoretical calculations [60]. It is noted that the prediction of a variety of measurable spectroscopic properties, including vibrational spectra and the molecular properties of the new systems, is particularly helpful for studying molecular conformation [43]. Therefore, computational spectroscopy is an invaluable tool for experimental spectra assignment and their interpretation in terms of basic physicochemical properties. Currently, modern computational chemistry yields results comparable to the most accurate experimental measurements [61]. Furthermore, the interpretation of experimental vibrational spectra is much easier and accurate owing to calculated harmonic and anharmonic [62] vibration transitions [63].
The experimental νC=O values with theoretically calculated frequencies [51] of some 2-acylpyrroles are summarized in Table 1. It was observed experimentally that 2-acylpyrroles under study form essentially doublet carbonyl bands. For some of the pyrroles the low frequency band of the carbonyl group has an asymmetric shape. It is a consequence of an association phenomenon since a concentration experiment removes this band asymmetry. Thus components of the doublet were determined to show characteristic C=O absorption of the free syn- and anti- forms [40-42]. The formation of associated forms was the cause of the asymmetry of the low frequency carbonyl band. It is noted that 2-substituted pyrroles tend to form dimeric hydrogen bonded species for steric reasons (Figure 2). Therefore, they exist as dimers which gives rise to an additional C=O band. This band is slightly red-shifted compared with the C=O band of the free syn or anti-form. This phenomenon is a result of weakening of the dimer C=O bond in comparison with the monomer species [24]. As the solution is more diluted the decrease in band intensity for the hydrogen bonded carbonyl group and increase in absorption intensity for the free C=O group was observed. In concentrated solutions 2-acylpyrroles exist as N-H…O=C dimers with consequent lowering of the carbonyl stretching frequency. For instance νCO for the anti and syn-form of ester 6 and 7 is 1719 and 1701 cm-1, respectively, while the νCO band for the hydrogen bonded carbonyl group is at 1689 cm-1 (0.3M CCl4) [42]. The IR spectra of aldehyde (2,3) in CCl4 and cyclohexane reveal single absorption of the free carbonyl group at 1666 cm-1, whereas absorption of the C=O group bonded by the hydrogen bond was observed at 1656 and 1659 cm-1, respectively [42]. It is noted that cyclohexane is the least interactive solvent and has one of the lowest dielectric constants (2.02). Therefore, cyclohexane is considered the paradigm of a non-polar solvent. Conformer abundances in this medium will thus be most similar to those in the vapor state. It is worth noticing that the theoretically estimated C=O frequency for the pyrrole-2- carbaldehyde dimeric species is 1668 cm-1 [60]. Similarly, IR bands of anti, syn and associated forms of the carbonyl group were observed for chloropyrroles (14-19,33-36) [41]. In the IR spectra, there are three absorption bands originating from the anti-conformer, syn-conformer and dimeric form [41]. There are absorption maxima at 1662, 1683 and 1646 cm-1 for 14, 15, 1658, 1683 and 1656 cm-1 for 16,17 and 1670, 1679 and 1657 cm-1 for 18,19. Theoretical calculations predict [41] that the νC=O band of the anti- form lies at a higher frequency than the νC=O band of the syn-form (Table 1). Having considered these calculations, it was anticipated that the νC=O band of the syn conformer was at a lower frequency and overlapped with the νC=O band of the hydrogen-bonded carbonyl group. The Fourier Self-Deconvolution (FSD) technique [64,65] was also applied to confirm this assumption. Based on these results the presence of stable syn and anti-conformers in solution was confirmed [42,60].
Recently, experiments in the gas phase, by microwave spectroscopy [66] and infrared imaging in supersonic jet expansion [25] also showed that aldehyde (2) adopts the syn-conformation. The anti-conformer has a high relative energy and its equilibrium population at room temperature was estimated [25] to be very low (1%). Fausto [67] has recently found that UV irradiation of matrix-isolated syn-pyrrole-2- carbaldehyde promotes internal rotation of the aldehyde CO group, leading to isomerization of the syn conformational ground state into the less stable anti-form. The anti-form of pyrrole-2-carbaldehyde was experimentally observed and characterized using low-temperature matrix isolation combined with IR spectroscopy.
Solvent interactions have also been used to study pyrrole-2- carboxylic acid and N-methylpyrrole-2-carboxylic acid conformers. Based on these studies [40], it was found that pyrrole-2-carboxylic acid and its N-CH3 derivative exist in solution in the most stable syn conformation. Because of poor solubility in non-polar solvents, the syn/anti ratio of pyrrole-2-carboxamide (8,9) was evaluated using 13C NMR on the basis of 1H decoupling without any Nuclear Overhauser Effect (NOE). The ratio was found to be 3:1 in DMSO solution [68].
Solvents of different polarity: It was found that conformational equilibrium depends on solvent polarity [69]. When the two conformers have very different dipole moments, spectra in solvents with different polarities reveal variable intensities of some bands. It was concluded based on the measurement of mono- and diesters of some N-alkylpyrroles (such as 2- or 2,3-dimethoxycarbonyl-N-ethyl-, -propyl-, -n-butyl- or -t-butylpyrrole) in polar and non-polar solvents that 2-monoesters of N-alkylpyrroles exist in solution in one syn-form only [57]. The greater stability of the syn over the anti rotamer in lowpolarity solvents is due to its lower dipole moment.
The study of 2-acylpyrroles reveals that there is only one carbonyl band in the spectra of 2-substituted N-methylpyrroles [57,59,60]. Our latest spectroscopic and theoretical studies show that two conformers of some chloropyrroles (33-36) are present in cyclohexane solution [41]. IR spectra revealed two carbonyl bands at 1658 and 1683 cm-1 for (35,36) and two νC=O bands at 1662 and 1683 cm-1 for (33,34) whereas only one carbonyl band at 1681 cm-1 was found for N-methylpyrrol-2- yl trichloromethyl ketone (38) [41,42]. These IR data were surprising because according to earlier investigations we could expect to find only one νCO band originating from the syn-form of N-methyl-2- acylpyrroles [70-73]. For instance, the IR spectrum of N-methylpyrrole- 2-carboxylic acid, run in a non-polar solvent, shows single absorption of the carbonyl group [40]. There are also other N-alkyl-2-acylpyrroles such as N-methyl-, N-ethyl-, N-propyl, N-butylpyrroles which exist in solution in only one, the most stable syn-form. It is worth noting that long chain N-substituted alkyl 2-acylpyrroles easily adopt the synconformation [71].
Infrared spectra of N-methyl 2-acylchloropyrroles (33-38) were also investigated in acetonitrile solution to distinguish the anti and the syn-rotamers [74]. It is noted that a method is available to discern the anti and the syn-rotamer. It was found that the position of equilibrium between alternative rotational isomers of a particular molecule is medium-dependent. Solvent plays a role in determining equilibrium between rotamers due to its influence on thermodynamic properties such as enthalpy, free energy, and total energy [75]. Owing to IR spectra recorded in different solvents changes could be observed in the position of the ester (6) carbonyl bands [41]. The variety of factors responsible for the observed differences include π-electron delocalization and vibrational coupling of normal modes. The greater stability of the syn form over the anti-rotamer for 2-acylpyrroles in low polarity solvent is due to its lower dipole moment. The moments for the pyrrole ring and the oxo-group are mutually opposing which leads to much lower dipole moments of the syn- forms [68]. The νCO bands of the more polar anti-form (36,34) were shifted to lower frequency compared to the C=O band position observed in cyclohexane. Furthermore, the AsynνC=O/AantiνC=O ratio of integrated intensities of the carbonyl bands for (33/34) is sensitive to solvent polarity. The ratio is smaller in acetonitrile solution compared to the value in less polar cyclohexane solution. It means that the population of the more polar anti-form increases with the increase in solvent polarity [75]. This effect was also observed for long chain N-substituted alkyl 2-acylpyrroles [71]. It would be expected on the basis of carbonyl absorption data that the N-H group could also show two absorption bands of the syn and anti-form spaced apart. Therefore, the studies were extended to include the νNH region. The vibrational assignment of the νN-H modes of 2-acylpyrroles (2-19) was facilitated using DFT calculations [41]. Since the difference between frequencies of νN-H is theoretically calculated depending on the spatial arrangement of the carbonyl group [24], it would be expected that a difference exists between the syn and anti νN-H frequencies in the experimental spectra. For instance, in the high frequency region of the IR spectra of methyl pyrrole-2-carboxylate two prominent bands at 3465 and 3318 cm-1 were observed [42]. It was noted that lower frequency band intensity considerably decreased as dilution increased, in favor of the band at 3465 cm-1. At the same time a reduction in the asymmetry of the C=O band was noted. The observed effects of concentration were rationalized assuming the formation of centrosymmetric dimers (Figure 2) [24]. Further confirmation of the formation of the dimer was provided by comparing experimental and theoretically calculated spectra of monomers (the syn- and antiform) and the dimeric form. The theoretical calculations confirmed assumption about the formation of the centrosymmetric dimers [24,42,76]. On this basis, the rotational isomers and a stable dimeric form were found in solution.
Based on the calculated energy of formation of the syn and antiform of methyl pyrrole-2-carboxylate [42], it was found that the synform was more favored relative to the anti-form in the gas phase (Table 1). The dramatic concentration dependence of absorption at 3318 cm-1 was explained by the breakage of intermolecular hydrogen bonds. Thus the band at 3318 cm-1 was assigned to νN-H of the hydrogen bonded form of ester (6). In addition, two slightly separated bands were detected in the N-H high frequency region. The separation phenomena of the free νN-H bands occurred in cyclohexane solution only. However, in solvents with higher dielectric constants separation of the free N-H bands of the syn and anti-form was not observed. It is noted that 2-acylpyrroles are mostly insoluble in non-polar media. Therefore, some IR experiments were performed in cyclohexane solution only. In other solvents, this effect was not observed. The spectrum of ester (6) recorded in cyclohexane revealed an N-H band at 3465 cm-1, assigned to the stretching vibration of the free N-H group which revealed a shoulder band around 3480 cm-1. This observation further confirmed that there were two conformers in the solution. The syn-form is expected to have a lower wave number since the calculated vibrational modes reveal such a tendency (Table 1).
Fourier self-deconvolution of IR spectra (FSD-IR): Overlapping bands may be separated using the Fourier self-deconvolution (FSD) method [77]. Fourier self-deconvolution (FSD) is a mathematical means for separating overlapped bands. There are many examples of how overlapped infrared spectra can be enhanced and greatly improve their information content. One of them is estimation of protein secondary structure and conformations by the analysis of the resolution-enhanced amide I profile by FSD [78]. Thus the FSD-IR was used to extract individual components from a composite band of N-H and C=O groups. In such a way separation of the overlapping bands was achieved, even though the spectral bands could not be resolved by collecting spectral data even at high resolution settings. The FSDIR method was applied for separating some overlapping bands of pyrrole-2-carboxylic esters [42]. Using this method the band νN-H at 3479, 3465 cm-1 was assigned to the anti and the syn-form, respectively. Considering the theoretical calculations which predicted that the νC=O band of the anti-form should be at a higher frequency than the νC=O band of the syn-form, we anticipated that the νC=O bands of the syn conformer might overlap with the νC=O band of the hydrogen-bonded carbonyl group. This FSD procedure allowed to identify the absorption band of the carbonyl group of pyrrol-2-yl dichloromethyl ketone (16) linked by hydrogen bonding [41].
Hydrogen bonds are considered as a factor that plays a very important role in many chemical and biological phenomena [79]. H-bonds can be classified according to atomic electronegativity criteria derived from a report by Desiraju and Steiner [80] and according to spectroscopic, structural and thermodynamic properties of H-bonds proposed by Jeffrey [81]. Jeffrey has noticed for the first time that some intercorrelated parameters such as H-bond energies, geometries, stretching frequencies of the proton donating group are sufficient to classify the interactions as strong, moderate and weak hydrogen bonds. Based on the above assumptions a number of experimental and theoretical methods aimed at the characterization of hydrogen bonds have been developed. It was observed that hydrogen bonds have a locking effect on single C-C bond conformations. For example, the most stable conformer of butane-2,3-diol has gauche orientation, which leads to the formation of an intramolecular hydrogen bond [82]. We can say that hydrogen bonds control the conformation of the molecule.
In this section the theoretical methods used to investigate 2-acylpyrrole hydrogen bonds are presented.
Theoretical studies of cyclic dimers
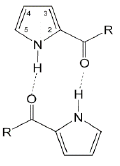
On the basis of spectroscopic investigations of pyrrole-2-carboxylic acid (4) using both IR and Raman techniques, it was revealed that pyrrole-2-carboxylic acid forms cyclic acid dimers (Figure 2) in the solid state. Theoretically estimated IR frequencies for the syn-dimers were very close to the experimentally observed wave numbers. It was thus found that the syn-conformation exists in the solid state as well as in solution [40]. Subsequently, the crystal structure of pyrrole- 2-carboxylic acid (4) determined by single-crystal X-ray diffraction proved that both C=O…H-O and N-H…O=C syn-dimers exist in the solid state [83].
These hydrogen bonding motifs are well known in the crystal structures of organic compounds. For example, the existence of motifs containing N-H…O bonds was observed for the crystal structure of methyl 5-methyl-3,4-diphenyl-1H-pyrrole-2-carboxylate [84]. Pyrrole-2-carboxylic acid (4) has a proton donating and proton accepting COOH carboxylic group as well as an N–H proton donating bond. All of them are involved in hydrogen bond interactions in the crystal structure of acid (4) where centrosymmetric dimers are formed with two equivalent O–H...O bonds between the carboxylic groups and N–H...O bonds between the N–H pyrrole ring and the C=O carbonyl group. Hence, according to Etter grafs’ designations, the following H-bond motifs were detected in the crystal structure of the acid: R2 2(8) and R2 2 (10).
To evaluate the strength of interaction, H-bond energies were calculated for pyrrole-2 carboxylic acid dimers at the B3LYP/6- 311++G(d,p) level of theory. Because the molecule contains O-H, C=O, and N-H groups it may form two types of hydrogen-bonded cyclic dimers. There are syn-dimers which form through C=O…H-O bonds and the cyclic dimer which forms two intermolecular interactions of the N-H…O=C type. The H-bond binding energies were computed as the difference between the total energy of the complex and the energies of the isolated monomers and further corrected for the basis set superposition error (BSSE) using the counterpoise method [85].
The energy of the H-bonds within the dimers (C=O…H-N) is of medium strength (Table 2): 5.9 kcal/mol. The H…O contact length is 1.880 Å, and the N-H…O angle is 163.7°.
 |
R=H | R=OH | R=CH3 | R=CH2Cl | R=CHCl2 | R=CCl3 |  |
R=NH2 R1=H | R=NH2 R1=CH3 | R=NHCH3 | R=OH | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| syn | anti | syn | anti | syn | anti | syn | anti | ||||||||||||
| B3LYP/6-311++G(d,p) | |||||||||||||||||||
| N-H | 1.024 | 1.020 (1.019) | 1.022 | 1.022 | 1.022 | 1.019 | 1.026 | 1.027 | 1.01 | 1.009 (0.996) | 1.008 (0.99) | ||||||||
| C=O dim | 1.231 | 1.225 (1.226) | 1.235 | 1.228 | 1.227 | 1.222 | 1.243 | 1.235 | 1.241 | 1.238 | 1.251 | 1.238 (1.237) | 1.231 (1.230) | ||||||
| H…O | 1.835 | 1.88 (1.892) | 1.855 | 1.855 | 1.847 | 1.897 | 1.865 | 1.872 | 1.857 | (1.697) | (1.688) | ||||||||
| ∠X-H..O X= N, O | 164.4 | 163.7 (163.9) | 161.1 | 163.2 | 164.2 | 164.6 | 178.6 | 172.7 | 179.9 | 179.7 (178.9) | 180.0 (178.6) | ||||||||
| ΔvC=O (vmon-vdim) | 17 | 21 | 34 | 28 | 25 | 10 | 25 | 7.5 | 15.6 | (53) | (60) | ||||||||
| EHB [kcal/mol] | -6.9 | -5.9 (-5.7) | -6.23 | -6.45 | -6.63 | -5.26 | -6.78 | -6.96 | -6.01 | -6.57 | -6.98 | -7.61 | -8.0 (-7.7) | -8.5 (-8.3) | |||||
| ρBCP [au] | 0.030 | 0.0263 | 0.0283 | 0.0278 | 0.0282 | 0.0261 | 0.0303 | 0.0298 | 0.0314 | 0.0483 | 0.0498 | ||||||||
| ρRCP [au] | 0.0029 | 0.0030 | 0.0036 | 0.0036 | 0.0036 | 0.0048 | 0.0049 | 0.0058 | 0.0081 (0.008) | 0.0081 (0.008) | |||||||||
| Reference | [58] | [39] | [40] | [40] | [40] | [40] | [69] | [69] | [88] | [88] | [39] | [84] | |||||||
Table 2: Geometrical, and topological parameters of X–H…O=C hydrogen bonds (in Å and degrees) with H-bond energies EHB in kcal mol-1 of 2-acylpyrrole dimers calculated at the B3LYP/6-311++G(d,p) level of theory; data calculated at B3LYP/6-311+G* are given in parentheses. The ring critical point was created due to double hydrogen bond formation through the C=O and N-H bonds or through C=O...H-X.
According to theoretical calculations dimers linked through two C=O…H-O bonds are more stable than those formed through N-H… O=C bonds. Additionally, the O-H…O bond within the dimer formed by the syn-conformers is stronger than the O-H…O bond within the anti-dimer [40,83]. EHB is 8.0 kcal/mol. The H…O contact length is 1.661 Å and the O-H…O angle is 179.7°.
The nature of HB interactions within the aldehyde (2) dimer was investigated [42] using theoretical calculations at the B3LYP/6- 311++G** level of theory. This study showed elongation (0.02 Å) of the C=O and N-H bonds for the syn-dimer of aldehyde (2) for which double connection through two N-H=O hydrogen bonds exists. The calculated energy of the single N-H…O interaction within the dimer is -6.9 kcal/mol [42]. The H…O contact length is 1.835 Å and the N-H…O angle is 164.4°.
The crystal structure of pyrrole-2-carboxamide (8) was also determined by single crystal X-ray diffraction [68]. It was found that the syn-conformer exists in crystals forming N-H…O bifurcated hydrogen bonds, since there is one accepting center (C=O) and three proton donors (N-H). For the crystal structure of pyrrole-2-carboxamide (8) there are centers of inversion within the eight-membered R2 2(8) rings [86] of the connected amide groups. It is noted that the same motif is found in compounds containing carboxylic groups, such as the syndimer of acid (4). There are also R2 2(10) H-bond motifs for the amide crystal structure.
DFT calculations were performed to analyze H-bonds in the amide dimers. N-H...O and O-H...N bonds in centrosymmetric dimers were taken into account with transition states corresponding to the N-H...O ↔N...H-O double proton transfer reaction. The geometrical and energetic features of such H-bonds show that these interactions may be classified as intermolecular resonance-assisted hydrogen bonds. The investigation of different tautomeric forms in dimers shows that OH…N bonds are stronger than NH…O bonds since the former are closer to the corresponding transition states. The calculated (B3LYP/6- 311++G(d,p)) strength for the N-H..O interactions within the amide syn-dimer is 13.57 kcal/mol [68]. It is stated that for acid and amide crystal structures H-bonds attributed to R2 2(8) motifs may be classified as intermolecular resonance assisted hydrogen bonds since the strength of these interactions is enhanced owing to π-electron delocalization [87].
The syn-conformer was also observed in the crystal structure of 1-methylpyrrole-2-carboxamide (27) [88] A centrosymmetric dimer connected through equivalent N–H…O hydrogen bonds was formed. There are R2 2(8) H-bonded motifs with two N–H...O hydrogen bonds equivalent owing to symmetry constraints. Such motifs are also observed for acid (4) and amide (8) crystal structures and are very common for the crystal structures of carboxylic acids and of amides.
B3LYP/6-311++G** calculations on centrosymmetric dimers of both N-methylpyrrole-2-carboxamide conformers connected through equivalent N–H...O hydrogen bonds were carried out. The calculated binding energy of the N-H…O intermolecular HB is 11.49 and 12.67 kcal/mol for the syn and anti-dimer, respectively.
For the crystal structure of 1-methylpyrrol-2-yl trichloromethyl ketone (37) there is only one typical proton accepting the carbonyl group and there are no typical proton donors. Therefore, only one type of hydrogen bond exists: -C–H...O. Due to the lack of typical proton donating bonds the pyrrole ring C–H bond acts as a proton donor. Additionally for the crystal structure of 37 the C–Cl...O halogen bond is found. For these interactions C–Cl bonds act as Lewis acids. In the crystal structure of 1-methylpyrrol-2-yl trichloromethyl ketone (37) C–Cl...O halogen bonds were found [70]. Furthermore, weak C–H…O hydrogen bonds exist. The C-Cl...O distance is 3.047 Å. This distance is slightly shorter than the corresponding sum of van der Waals radii. This means that, similarly as H-bonds, the halogen bonds may also influence the arrangement of molecules in crystals. There is also a short C4–H...O contact of 2.64 Å. It is noted that that the C–Cl…O halogen bond forms a repeated chain motif: …O=C–CCl3 …O=C–CCl3 … according to the crystal translational symmetry. This is a so called cooperative effect well known for H-bonds in crystal structures. It is also stated that in the crystal structure of 1-methylpyrrol-2-yl trichloromethyl ketone the syn-form exists.
The analysis of the crystal structure of some 2-acylpyrroles shows that the energetically more stable syn-form is present. For all of the above crystal structures of 2-acylpyrrole derivatives the carbonyl group plays a crucial role influencing the arrangement of molecules in crystals. For the crystal structure of pyrrole-2-carboxylic acid (4) the syn-conformation exists; the same syn-conformer is observed for the pyrrole-2-carboxamide (8) crystal structure. The crystal structure of N-methylpyrrole-2-carboxamide (27) also shows the existence of the syn-conformation.
The crystal structure of the acid (4), and amide (8,27) dimer reveals presence of centrosymmetric syn-dimers (Figure 2). Stronger H-bonds were detected for the dimers connected by C=O…H-O bonds compared with those with N-H...O=C bond. For the syn-dimers of acid (4) intermolecular RAHBs exist, whereas N-H…O=C dimers are not assisted through resonance, since the interactions (N-H…O=C) are not supported by the existence of effectively mixing tautomeric forms.
Heteronuclear intermolecular hydrogen bonds are found in the crystal structures of the amide (8) dimer connected by C=O…H-N. Heteronuclear interactions are usually much weaker than homonuclear H-bonds, since there is a difference between proton affinities of the oxygen and nitrogen centers. The calculated EHB for the syn-amide (8) dimer is 6.78 kcal/mol, whereas for the syn-acid dimer (4) it is 8 kcal/mol. Intermolecular interactions C=O...H-N for the syn-Nmethylamide dimer (27) are weaker (6.01 kcal/mol) compared with the EHB of dimers (4,8). An effect of the equalization of C-O and C=O bonds within the carboxylic groups in the crystal structures of acid dimers (4) was observed. However, for amide dimer (8) the effect was less evident due to the existence of weaker C=O...H-N interactions. Furthermore, it was observed that HB binding energies corresponding to the antidimers were greater than EHB interactions for the syn-counterparts. This phenomenon is related to the Leffler–Hammond postulate that systems with stronger H-bonds are closer to transition states and hence they have greater energies [89]. It was concluded based on theoretical calculations that binding energy depends largely on the conformer type (Table 1).
AIM studies
Other commonly Accepted Date: criteria for the description of hydrogen bonds were introduced by Koch and Popelier [90]. They proposed that hydrogen bonds can be characterized based on various descriptors originating from Bader’s quantum theory of “Atoms In Molecules” (AIM) [91]. There are eight effects for charge density that are the necessary criteria for the existence of inter- or intramolecular hydrogen bonds. Among them there are the bond critical point (BCP) for H…Y (where Y is a proton-acceptor center within the X-H…Y hydrogen bond) and the ring critical points (RCP) existing due to the formation of an intramolecular hydrogen bond or due to the formation of a dimer. The value of electron density at the BCP (ρH. . Y) should be within a range of 0.002–0.040 a.u. The corresponding Laplacian of electron density at the BCPs (∇2ρBCP) should be within a range of 0.024-0.139 a.u. Additionally there are energetic descriptors of BCPs such as electron energy density at BCP (HC) and its components, potential electron energy density (VC) and kinetic electron energy density (GC). According to Rozas et al. [92] weak H-bonds are characterized by ∇2ρBCP>0 and HBCP>0. It is noted that weak hydrogen bonds are predominantly electrostatic and the distance between the interacting atoms is less than the sum of their van der Waals radii. Topological analysis of the above electron density at the BCP clearly reflects the properties of intermolecular or intramolecular contacts [93,94]. Besides, the quantum theory of “atoms in molecules” QTAIM enables studying hydrogen bonds independently from spectroscopic, X-ray diffraction, and classical physicochemical techniques.
Theoretical studies of the nature of hydrogen bond interactions are still the subject of many reports since they play a key role in various chemical, and biochemical processes. It was revealed that electron density at the H-bond BCP may be treated as a measure of hydrogen bond strength [95-98]. The parameters of the ring critical point RCP (Figure 4) may also be considered as descriptors of hydrogen bond strength [99]. Correlations between the characteristics of RCPs and other parameters describing H-bond strength were observed by Alkorta [100,101]. There is also a good correlation between electron density at BCPH…Y and H-bond energy for pyrrole-2-carboxylic acid dimers [40]. It was observed that the stronger the H-bond, the greater the ρH…Y value. This effect was most evident for a homogeneous set of samples such as pyrrole-2-carboxylic acid (4,5) dimers, [40] and pyrrol-2-ylchloromethyl ketone (14,16,18) dimers (Table 2) [41].

Figure 4: Molecular graphs of the systems investigated. Big circles correspond to attractors attributed to nuclei and small circles to critical points, bond critical points (BCPs), and ring critical points (RCPs).
Linear relationships with good correlation coefficients (0.998) were found between the H…O intermolecular distance and the ρH…O value for the pyrrole acid dimers and related complexes [83]. Similar correlations between the geometrical and topological parameters were observed recently for different samples of H-bonded systems, such as syn- and anti-1,1-di(pyrrol-2-yl)-2-formylethene [4].
A linear relationship between C=O bond length and electron density at the corresponding bond critical point ρBCP was observed for the 1-methylpyrrole-2-carboxamide (27) dimer as well as for analogous systems such as 1-hydroxypyrrole-2-carboxamide [102].
There are also correlations between C=O bond length and Laplacian of the electron density at the BCP and total, kinetic and potential electron energy densities at this BCP (HC, GC and VC) 1-methylpyrrole-2-carboxamide (27) [88].
AIM methodology has been applied to exclude or confirm the existence of intramolecular hydrogen bonds [102-106]. In an earlier study of the spectroscopic properties of pyrrole-2-carbaldehyde, the formation of intramolecular interactions C=O…H-N was proposed [30]. However, the analysis of structural parameters obtained by X-ray diffraction and theoretical calculations as well as spectroscopic measurements, revealed that there are no intramolecular hydrogen bonds in the 2-acylpyrroles [42]. Additionally, the Quantum Theory of Atoms in Molecules (QTAIM) was applied to study the possibility of intramolecular C-H...O=C hydrogen bond formation in pyrrole-2- carbaldehyde (1) and its derivatives. The analysis of the bond critical points shows that there is no intramolecular hydrogen bond for the syn conformer of aldehyde (1) or for the ester (5). For these molecules it was not possible to find the bond critical points corresponding to the intramolecular N-H...O contacts [24,42]. Figure 4 presents two examples of molecular graphs of the 2-acylpyrrole dimers. The complexes are linked through N–H...O interactions. The H-attractor of the proton donating N–H bond is linked through the bond path with the BCP of the C=O bond. Topological analysis shows that there are no intramolecular hydrogen bonds for the syn-conformer of pyrrole-2- carbaldehyde (2) or for methyl pyrrole-2-carboxylate (6).
In search for a good method for describing hydrogen bond strength, electron density at the ring critical for pyrrol-2-yl-chloromethyl ketone (14,16,18) dimers was considered as a descriptor of hydrogen bond strength. Figure 2 presents small circles corresponding to ring critical points. The difference between ρRCP and ρBCP (N-H…O) characteristics for these dimers was not meaningful. It means that electron density at the ring critical point ρRCP and the bond critical point ρBCP are not sensitive to the R substituent [41].
X-ray diffraction
This chapter presents the results of X-ray studies of some 2-acylpyrroles and the structural information available in the Cambridge Structural Database (CSD) [107,108].
Data analysis includes 2-acylpyrrole dimers connected by two N-H…O=C hydrogen bonds. For these dimers there are R2(10) motifs containing two equivalent N-H...O=C hydrogen bonds. The intermolecular C=O…H-N interactions are the subject of numerous studies since they play a role in many chemical and biochemical processes. Spectroscopic and theoretical studies on the conformations of some 2-acylpyrroles reveal that the syn-conformer, in which the carbonyl group is located on the same side as the N–H or N–CH3 group, is more stable than the anti-form. Consequently, such a conformation of the molecule favors the formation of centrosymmetric dimers with two equivalent intermolecular hydrogen bonds as shown in Figure 2. The most important experimental methods which provide reliable information concerning the spatial arrangement of the atoms in a molecule are diffraction methods. These methods include X-ray and neutron diffraction to retrieve information about molecules such as bond lengths, bond angles, torsional angles and the position of the hydrogen atom in the hydrogen bond. The later data indicate the existence and strength of the interaction. The primary information on hydrogen bonds from diffraction studies is the N-H…O and C=O distance, and elongation of the N-H bond. Most of these data concern the analysis of intermolecular H-bonds in 2-acylpyrroles. Valuable information from diffraction analysis of 2-acylpyrroles was reported by Grabowski and co-workers [68,70,83,88]. The authors assessed the N-H...O and O-H...O interaction on the basis of crystal structure, IR analysis and theoretical calculations. Structural studies and theoretical analysis of HB interactions pointed to the formation of centrosymmetric dimers with two equivalent intermolecular N-H...O hydrogen bonds. Other data are also available on the crystal structure of 2-acylpyrroles. Cambridge Structural Database (CSD) searches on 2-acylpyrroles reveal that there are also some other instances of 2-acylpyrroles which form dimers in the solid phase by N-H…O=C interactions [109-125]. Table 3 presents (N)H…O, N…O and C=O bond lengths of selected 2-acylpyrrole dimers bonded by hydrogen bonds.
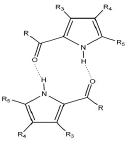 |
(N)H...O=(C) | dN...O | dC=O | Reference |
|---|---|---|---|---|
| [Å] | ||||
| 4-Chloro-2-formyl-3-methyl-pyrrole R=H; R3=CH3; R4=Cl; R5=H |
1.904 | 2.807 | 1.222 | [110] |
| 2-Formyl-3-methyl-pyrrole R=H; R3=CH3; R4=; R5=H |
1.883 | 2.806 | 1.234 | [110] |
| 3-Methoxypyrrole-2-carbaldehyde R=H; R3=OCH3; R4=H; R5=H |
1.816 | 2.797 | 1.223 | [111] |
| Methyl 2-Formylpyrrole-3-carboxylate R=H; R3=COOCH3; R4=H; R5=H |
1.966 | 2.842 | 1.226 | [111] |
| N,N-Dimethyl-1H-pyrrole-2-carboxamide R=N(CH3)2; R3=H; R4=H; R5=H |
1.909 | 2.817 | 1.246 | [112] |
| Methyl 5-methyl-3,4-diphenyl-1H-pyrrole-2-carboxylate R=OCH3; R3=C6H5; R4= C6H5; R5=CH3 |
1.99 | 2.829 | [85] | |
| Diethyl 5-methylpyrrole-2,3-dicarboxylate R=OC2H5; R3= OC2H5; R4= H; R5=CH3 |
1.911 | 2.933 | 1.214 | [113] |
| Ethyl 4-dodecyl-3,5-dimethyl-1H-pyrrole-2-carboxylate R=OC2H5; R3= CH3; R4= C12H25; R5=CH3 |
1.995 | 2.85 | 1.212 | [114] |
| 4-Ethyl-3,5-dimethyl-1H-pyrole-2-carboxylic acid ethyl ester R=OC2H5; R3= CH3; R4= C2H5; R5=CH3 |
2.036 | 2.86 | 1.224 | [115] |
| Ethyl 4-acetyl-3,5-dimethyl-1H-pyrrole-2-carboxylate R=OC2H5; R3= CH3; R4= COCH3; R5=CH3 |
2.00 | 2.86 | 1.216 | [116] |
| 2-Trifluoroacetyl-5-trifluoromethylpyrrole R=CF3; R3= H; R4= H; R5=CF3 |
2.015 | 2.860 | 1.219 | [117] |
| Pyrrole-2-carboxylic acid R=OH; R3= H; R4= H; R5=H |
2.18 | 2.997 | 1.23 | [84] |
| Dimethyl 3,5-dimethylpyrrole-2,4-dicarboxylate R=OCH3; R3= CH3; R4= OCH3; R5=CH3 |
2.08 | 2.863 | 1.219 | [118] |
| Pyrrole-2-carboxamide R=NH2; R3= H; R4= H; R5=H |
2.207 | 2.987 | 1.249 | [69] |
| 1-(2-Pyridyl)-1-(2-pyrrolyl)methanone R=C5NH4; R3= H; R4= H; R5=H |
2.045 | 2.86 | 1.238 | [119] |
| 2-Acetylpyrrole R=CH3; R3= H; R4= H; R5=H |
1.983 | 2.863 | 1.227 | [120] |
| 4-Iodo-1H-pyrrole-2-carbaldehyde R=H; R3= H; R4= I; R5=H |
1.996 | 2.842 | 1.216 | [121] |
| Ethyl 5-formyl-3,4-dimethyl-1H-pyrrole-2-carboxylate R=OC2H5; R3= CH3; R4= CH3; R5=CHO |
2.07 | 2.919 | 1.193 | [122] |
| Dimethyl 3,4-diiodo-1H-pyrrole-2,5-dicarboxylate R=OCH3; R3= I; R4= I; R5= COOCH3 |
2.114 | 2.936 | 1.195 | [123] |
| (4-Fluorophenyl)(1H-pyrrol-2-yl)methanone R=C6H4I; R3= H; R4= H; R5= H |
2.056 | 2.865 | 1.236 | [124] |
| 4-Ethyl-3,5-dimethyl-1H-pyrrole-2-carbaldehyde R=H; R3= CH3; R4= C2H5; R5= COOCH3 |
2.085 | 2.889 | 1.226 | [125] |
| Phenyl(3,4,5-trimethyl-1H-pyrrol-2-yl)methanone R=C6H5; R3= CH3; R4= CH3; R5= CH3 |
2.034 | 2.890 | 1.24 | [126] |
Table 3: The hydrogen-bonding geometry [Å] of the 2-acypyrrole dimeric aggregates in solid phase. Data were taken from the structural information collected in the Cambridge Structural Database (CSD).
Among the dimers listed the mean d(N)H...O distance is 2.018 Å and differs generally by 0.05-0.1 Å. The mean dN...O distance is 2.875 Å and differs generally by 0.1 Å while the mean dC=O bond length is 1.22 Å. It is noted that the longest the C=O bond was observed for compounds in which R is NH2, OH, N(CH3)2. The shortest dC=O was observed for electron-accepting substituents such as R = OCH3, OC2H5. It was observed earlier that other electron-accepting substituents, such as R2 =COCCl3, COCHCl2, COCH2Cl, play a role in the stabilization of ring systems [51]. The more electron-withdrawing substituents, the greater π-electron delocalization and aromatic character expressed by the HOMA index of aromaticity [126].
Grabowski et al. observed hydrogen bonding in pyrrole-2- carboxylic acid using X-ray methods [83]. There are two symmetryequivalent N-H...O hydrogen bonds connecting the pyrrole rings with the carbonyl groups. Two symmetry-equivalent O...H-O bonds between the carboxylic groups were also found. These HB motifs were well suited to model dimers analyzed by applying infrared and Raman spectroscopy and DFT calculations (B3LYP/6-311+G(d)) [40]. A comparison of the calculated C=O length [83] shows that complexation causes equalization of C=O and C-O bond lengths when the carboxylic group participates in hydrogen bonding interactions. Therefore, it was concluded that the complexation process of pyrrole-2-carboxylic acid affects π-electron delocalization for connected species and the strength of hydrogen bonds. The C-H...O- H bonds have also been found in the pyrrole-2-carboxylic acid crystal structure. Hence, two equivalent O-H...O hydrogen bonds of the carboxylic groups are assisted through additional C-H...O and N-H...O contacts.
The bonding energy of dimers was also analyzed by Grabowski and co-workers using the Bader theory (“Atoms in Molecules” theory, AIM) [91]. It confirms that the H-bonds for the C=O...H-O dimer are stronger than those for the C=O...H-N dimer. The data in Table 2 show the quantitative parameters of the N-H...O=C, O-H...C=O HBs for the pyrrole-2-carboxylic acid dimers. It was recently reported that the H-bonds in pyrrole-2-carboxylic acid dimers are stronger than those in the dimers of formic or acetic acids. This phenomenon is most likely due to the charge-delocalization contribution to the interaction energy of the O-H...H double H-bonds [127,128].
The crystal and molecular structure of pyrrole-2-carboxamide was also analyzed using single crystal X-ray diffraction, FT-IR and NMR techniques, and DFT calculations [68]. The existence of two tautomeric forms for the dimers differing in H-bond motifs, N-H...O or O-H...N, was not found. The geometrical and energetic parameters of the hydrogen bonds show that these interactions may be classified as intermolecular resonance-assisted hydrogen bonds. It was also observed that the oxygen atom which acts as an acceptor center within the N-H...O H-bridge forms a bifurcated hydrogen bond. There is one accepting center (oxygen) and three proton donors. Three H-bond motifs corresponding to three HB connections are as follows: R2 2(8), R2 2(10) and R4 2(8) according to the designations devised by Etter and Bernstein [88]. According to chemical calculations the strongest N-H...O hydrogen bonds are formed through the amide groups. DFT calculations and AIM analyses indicate that the N-H...O hydrogen bonds may be classified as heteronuclear resonance-assisted hydrogen bonds (RAHBs).
The analysis of the crystal structure of some 2-acylpyrroles leads to the conclusion that the energetically more stable syn-form is present in the solid state. For all of the above crystal structures of 2-acypyrrole derivatives there is the crucial role of the carbonyl group hydrogen bonds influencing the arrangement of molecules in crystals. For the crystal structure of pyrrole-2-carboxylic acid (4) the syn-conformation exists. The same syn-conformer is observed for the pyrrole-2-carboxamide (8) crystal structure. The crystal structure of N-methylpyrrole-2- carboxamide (27) also shows the existence of the syn-conformation. The analysis of crystallographic data of 2-acylpyrroles indicates that the hydrogen bonds have a locking effect on conformation about the C-C bonds. The most stable form is the syn-conformer since it enables formation of a hydrogen bond between the pyrrole N-H and C=O groups.
Taking these results into consideration it seems that 2-acylpyrroles for which two conformers exist in solution as a rule crystallize exclusively in the most stable syn-conformation.
In this review, several examples showing how experimental (spectroscopic) and theoretical methods could be used to explore the conformational preferences of 2-acylpyrroles are presented. The most important method which was employed in the conformational studies of 2-acylpyrroles was infrared spectroscopy. This technique includes measurements in solution to identify the observed vibrational bands of one or more conformers. This method gave satisfactory results because IR spectroscopy is fast relative to the interconversion of 2-acylpyrrole conformers, hence the observed IR spectrum in solution is a weighed sum of the conformer spectra.
A comparative assessment of N–H…O=C hydrogen bonding interactions based on the author’s studies and on data available in the Cambridge Structural Database (CSD) is presented. Among the molecular properties HB energy, structural characteristics and topological parameters of electron densities at the BCP are particularly informative for the role of a substituent adjacent to the carbonyl group. Based on FT-IR and theoretical calculations, the C=O vibrational mode was found to be useful as a diagnostic tool for the analysis of hydrogen bond strength within 2-acylpyrrole dimers.
The theoretical calculations yielded conformational energies, potential functions for internal rotation, rotational barriers, electric dipole moments, and vibrational spectra. These results were compared with the available experimental data, and analyzed to identify the main factors determining the favored conformations of 2-acylpyrroles. 2-Acylpyrroles prefer the syn-conformation in the solid state and may be present in both the syn and the anti-form in solution. Theoretical calculations indicate the influence of syn and anti-conformations on π-electron delocalization. The electron-donating substituent groups R, such as NHCH3, N(CH3)2, NH2, OH, and OCH3, decrease π-electron delocalization, whereas the electron-withdrawing groups, such as CH2Cl, CHCl2, and CCl3, increase π-electron delocalization compared to the unsubstituted pyrrole ring. This phenomenon affects the strength of hydrogen bonds N-H…O=C. The stronger the electron-withdrawing property of the R group, the stronger the hydrogen bond within centrosymmetric dimers connected by N-H…O=C bonds.
For the acid (4) dimer connected by two C=O…O-H hydrogen bonds Table 2 intermolecular resonance-assisted hydrogen bonds (RAHBs) were detected. The real molecular structure of the pyrrole- 2-carboxylic acid dimer may be treated as a mixture of tautomeric forms. Their effective mixing was supported by the process of -electron delocalization. The geometrical and energetic features of H-bonds for pyrrole-2-carboxamide (8) dimers connected through the amide groups indicate that these interactions may be classified as heteronuclear intermolecular resonance-assisted hydrogen bonds.
Financial support from the National Science Centre is gratefully acknowledged (2011/03/B/ST5/02691).
Calculations were carried out in the Warsaw (ICM) Supercomputer Center (G53-7).