Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 8
Sucrose synthase is a key regulatory protein, and a potential biomarker for abiotic stress response in plants. These metabolic enzymes have been extensively examined for their varied functional roles. The upregulation of this Pi- and adenylate-independent glycolytic enzyme in different environmental contexts has also been extensively characterized. Here we present an analysis of the evolutionary features of sucrose synthases in an effort to correlate variations in sequence to the structure and function of this protein and its potential implication for the stress response mechanism in plants as evidenced from transcript distribution studies. An analysis of the evolutionary characteristics of this enzyme is useful given the role of this protein in sugar sensing, sugar import to sink tissues and plant development. Based on the secondary structural features of sucrose synthases and mapping of sequence variations in the context of the fold of these proteins, we note evolutionarily conserved amino acids of potential functional significance. In addition, we also observe the presence of target peptides in some of these proteins suggesting potential cellular localization. The results presented in this manuscript could aid ongoing studies on this protein as a potential biomarker and candidate gene for environmental stress.
Keywords: Sucrose synthase, Abiotic stress, Evolution, Target peptide.
Sucrose metabolic enzymes – SUS (sucrose synthase), SPS (sucrose phosphate synthase) and SPP (sucrose phosphatase) are ubiquitously expressed in cyanobacteria, proteobacteria and green plants. Homologues to sequences of known sucrose synthesizing enzymes exist in the α,β, and γ subdivisions of proteobacteria but not in other bacteria or archaea. It is reported that they could have been acquired by plants during the endosymbiosis of a sucrose synthesizing mitochondrial ancestor more likely of cyanobacterial origin since sucrose synthesis occurs in green plants with oxygenic photosynthesis. Sucrose synthesis is often known to occur in response to abiotic stresses such as salt or osmotic stress and is thought to help maintain membrane structure and function (Reed et al., 1986; Hagemann and Marin, 1999). Sucrose accumulation also occurs as a response to low temperature (Strand et al., 2003) or drought (Yang et al., 2001). Sucrose generally serves as a transport compound, shuttling carbon and energy between cells in filamentous species as well as higher plants and is a component of cell wall synthesis. Sequences of enzymes responsible for sucrose metabolism share common protein domains – sucrose synthase and glycosyltransferase domains, and the enzymes belong to the GT4 family in the carbohydrate active enzyme database (http://www.cazy.org/fam/GT4.html). Sucrose synthase activity correlates with sugar import (Sung et al., 1989), cell wall synthesis (Chourey et al., 1991) and sink strength (Sun et al., 1992). Sucrose synthase is involved with starch and sucrose metabolism; carrying out degradation (ADP/ UDP + sucrose = ADP-glucose + fructose) as well as synthesis (NDP-glucose + D-fructose = NDP + sucrose) of sucrose, with a characteristic choice of substrates viz., UDP>dTDP>ADP>CDP>GDP for the cleavage reaction. The pathway of sucrose degradation by sucrose synthase is favored particularly under energy limiting conditions because of lower overall energy costs, reflected by the regulation of the enzyme under oxygen deprivation. Sucrose synthase exists predominantly in the cytosol but is sometimes associated with the plasma membrane (Amor et al., 1995). Sucrose synthase is also noted to exist in a membrane bound form in association with cellulose or callose contributing to cell wall biosynthesis (Subbaiah et al., 2001). The enzyme is predicted to be a component of the mitochondrial proteome in maize that suggests roles other than sucrose catabolism (Subbaiah et al., 2006). This enzyme has also been reported to regulate nutrient/sugar signaling through the opening of mitochondrial permeability transition pores (Azoulay-Zohar, 2003). Besides the mitochondrion, sucrose synthase is also reported to be present in chloroplasts suggesting its role in regulating photosynthetic processes in plastids (Gerrits et al., 2001; Oswald et al., 2001).
There is a considerable body of literature on sucrose regulation and the crucial role of these metabolic enzymes in the functional biology of plants. However, important aspects of this metabolic process are still an enigma. Studies in cyanobacteria and the recent publication of the sequences of several complete genomes provide new insights into the origin and evolution of proteins involved in sucrose metabolism. It is difficult to reconcile these observations with previous phylogenic studies due to several isoforms and multiple sucrose metabolism gene transcripts. For example, in kiwifruit and wheat several protein isoforms exist as a result of polyploidization (Lunn and McRae, 2003). In Arabidopsis, there are at least six sucrose synthase genes and at least 3 SUS genes in citrus (Komatsu et al., 2002). The earliest phylogenetic studies of sucrose metabolic enzyme sequences using sequence information available at that time suggested that sucrose synthase genes could be classified into at least 3 major groups; one monocot and two dicot categories (Sturm et al., 1999). Alternative splicing of SUS transcripts have also been noted to be tissue or stage specific and thus constitutes a potential regulatory strategy. In maize, SUS1, SUS2 and SH1 are the three genes known along with isoforms. Some isoforms of these genes are known to be more responsive to stress, and mutational analysis suggests that this response may be paralog specific (Subbaiah et al., 2006).
Here we report our analysis of sucrose synthase protein sequences, their phylogeny and structural conservation and examine if there is a significant over-representation of sucrose synthase sequences in cDNA libraries derived after abiotic stress. This analysis builds on previous compilations of the SPS, SUS and SPP enzymes (Langenkamper et al., 2002; Lunn and McRae, 2003; Castleden et al., 2004; and Lunn, 2002; Komatsu et al., 2002; Subbaiah et al., 2006). The information generated through this study is thus likely to serve as a useful starting point to connect molecular events contributing to the stress response and the phylogenetic clustering of orthologs of genes involved in sucrose metabolism.
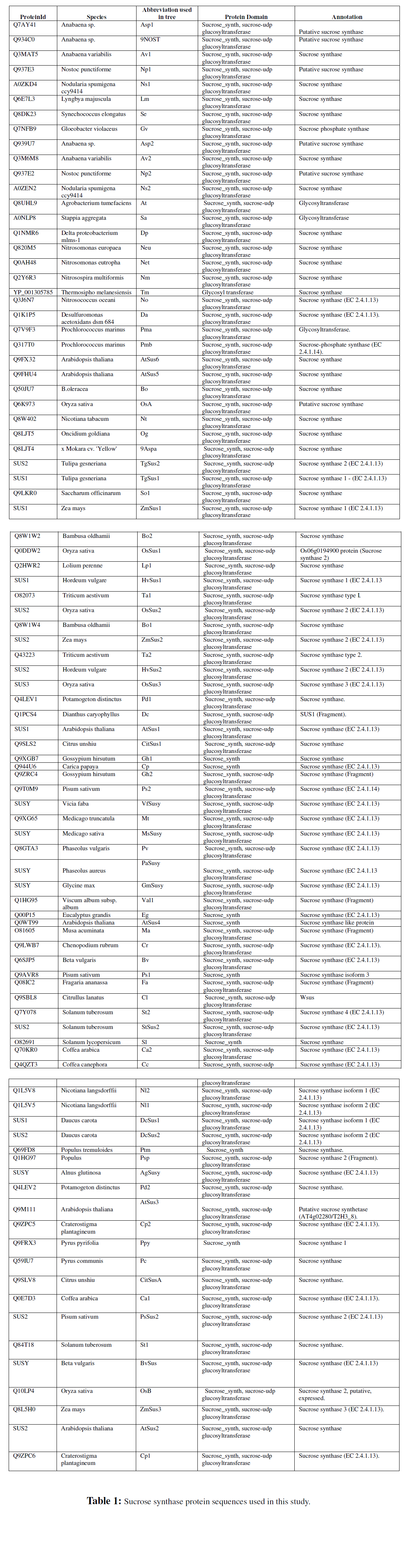
Sequence collection: Protein sequences corresponding to sucrose synthases were retrieved from the Pfam database (http://www.sanger.ac.uk/Software/Pfam/). A total of 160 sequences were recovered. Redundancy was checked using the standalone Blast program against the same dataset; isoforms were identified from a visual analysis of their alignments and validated using the isoform recognition tool IsoSVM (Spitzer et al., 2006).
Sequence analysis: An alignment of SUS sequences was performed using conventional software including ClustalW (Thompson et al., 1994), MUSCLE and PROMALS 2.0(Pei and Grishin, 2007). Alignment editing and visualization was carried out in Jalview(Clamp et al., 2004). Secondary structure prediction results were obtained from Promals that uses PSIPred as well as Jnet. Functional region identification through surface mapping was carried out using the Consurf server (Glaser et al., 2003). Phylogenetic tree construction was carried out using the PAUP4.0 software (Sinauer Associates). Both neighbour joining and parsimony trees were derived, using 1000 replicates. The representative alignments and trees shown in the paper were derived using ClustalW and PAUP software. The TreeView software was used to display trees (Page, 1996).The presence of N-terminal pre-sequences viz., mitochondrial targeting peptide (mTP) or secretory pathway signal peptide (SP) were determined using the TargetP 1.1 server (http://www.cbs.dtu.dk/services/TargetP/ , Emanuelsson et al ., 2000, 2007). User defined settings were selected for TargetP to indicate the plant origin of the sequences with “no cut-off” stringency for all sequences tested. The average amphipathicity, hydrophobicity and similarity plots were derived using AveHas (Zhai and Saier, 2002) with the input multiple sequence alignment generated using ClustalW. Protein motifs were searched using the PPsearch module that screens Prosite patterns (www.ebi.ac.uk/ppsearch). The reference sequences for each group of the phylogenetic tree were derived using Promals software (Pei and Grishin, 2007) and were searched against EST sequence databases at JCVI (formerly TIGR) using WU-Blast and separately against ESTs derived from abiotic stress cDNA libraries using a standalone version of BLAST(Altschul et al., 1997). The stress sequences were retrieved from an inhouse stress transcript database (http:// www.intranet.icrisat.org/gt1/tog/homepage.htm), which houses updated sequence data on stress derived ESTs across 16 crop species. The parsing of the BLAST output was carried out using scripts available in-house. Tree reconciliation was carried out using the Notung tool (Chen et al ., 2000) with species trees derived using the NCBI taxonomy browser.
Sucrose synthase is homologous to sucrose phosphate synthase, which catalyses the penultimate step of sucrose synthesis. The main sucrose metabolism enzymes viz., SUS, SPS and SPP differ in their domain architecture. The SUS sequences adopt two architectures: fusion of sucrose synthase and glycosyl transferase or the sucrose synthase domain alone. There are three variants to the SPS sequences: glycosyl transferase and sucrose phosphate synthase domains, sucrose phosphate synthase alone and a three domain structure with the sucrose synthase, glycosyl transferase and sucrose phosphate synthase domains. The SPP sequences have two architectures: a two domain S6PP with S6PP_C and S6PP alone. This study is based on sequences that display the SUS architecture. A search of the Pfam database resulted in 126 sequences containing the sucrose synthase and glycosyl transferase domain and 34 sequences with a lone sucrose synthase domain. These included 17 sequences of cyanobacterial origin, 9 of proteobacterial origin, one from the thermophilic anaerobic bacterium Thermosipho melanesiensis and 133 sequences from plants. The plant sequences consisted of 84 dicotyledonous representatives and 49 from monocots. After removal of redundant sequences, the total dataset was reduced to 135 sequences. Fragments of sequences less than 200 amino acids in length as well as isoforms were not retained for the phylogenetic analysis. Homologs from the same species whose alignment had sequence substitutions were retained to allow for more information content in the phylogenetic analysis. This dataset for the derivation of phylogenetic trees thus consisted of 71 plant sequences and 23 sequences derived from proteobacteria and cyanobacteria.
Phylogenetic Analysis
Figure 1a shows a phylogenetic tree with clear separation of the plant from the bacterial sequences. This is in agreement with earlier reports regarding cyanobacterial and plant SPS sequences (Langenkamper et al.,2002). Figure 1b shows a parsimony tree of plant sucrose synthase sequences and their separation into at least three sub - families. These three families have been labeled A, B and C. Of the 71 plant sequences, 20 monocot sequences clustered within sub-family A, 35 dicot sequences within a large sub-family B and 16 dicot and monocot sequences within sub-family C. All three sub-families are characterized by the presence of paralogues within a single species. Early classification of sucrose synthase indicated 4 main classes, class I consisting entirely of enzymes from monocots, class II, III and IV consisting entirely of enzymes from dicots (Komatsu et al., 2002, Sturm et al., 1999). Studies on SPS sequences reported clustering of SPS sequences into at least three families and every plant had a representative of each family in their genome. The clear distinction between dicot and monocot proteins is seen in our study as well; however dicot derived sequences are distributed into at least two subfamilies, one of which has monocot representatives. The phylogenetic trees generated using both Neighbour Join and Parsimony methods were in agreement and substantiated this clustering. The separation of A, B and C subfamilies is supported by bootstrap values of 88, 97 and 100 respectively. Within the subfamilies A and C, bootstrap support for all nodes range from 70-100 across the family, whereas within the dicot sub-Family B, there appear to be further divisions separating the family into at least 3 groups with bootstrap support of 77, 100 and 62. The clustering also appears to correlate with taxonomic separation, with representatives of the order Caryophyllales (Dianthus, Chenopodium and Beta vulgaris) clustering away from the Asterids sequences (Coffea, Nicotiana and Solanum) which, in turn, are separated from all the other representatives of the Rosids subclass (that includes Arabidopsis, Eucalyptus, Gossypium, Populus, Citrullus, Citrus and legume representatives). The dicot species that are represented in both Families B and C are Arabidopsis, Citrus, Pisum, Beta, Coffea and Solanum sp. The Arabidopsis representatives include six SUS proteins – SUS1 to SUS6 (Baud et al., 2004). SUS1 and SUS4 cluster with Family B, while SUS2, SUS3, SUS5 and SUS6 in Family C. There are some dicot sequences that do not have a homolog in B but are represented in sub-family C.

Figure 1: Phylogenetic trees and reconciled trees. (1A) Parsimony tree derived using PAUP software, showing the separation of plant sucrose synthase protein sequences from their cyano and proteobacterial ancestors.Labels correspond to the following sequences: 1-AtSus5,2-OsA,3-AtSus6,4-Cp1,5-Cp2,6-St1,7-AtSus4,8-Ppy,9-CitSusA,10-BvSus,11-AtSus2,12- OsSus2,13-Pd1,14-Ma,15-Og,16-Ta1,17-Pd2,18-AgSusy,19-DcSus1,20-Cl,21-Gh1,22-Val1,23-AtSus1,24-AtSus3,25- DcSus2,26-Ca1,27-Pc,28-PsSus2,29-OsB,30-ZmSus3,31-Bo1,32-ZmSus2,33-HvSus2,34-Ta2,35-OsSus3,36-9Aspa,37- TgSus2,38-TgSus1,39-HvSus1,40-Lp1,41-Bo2,42-ZmSus1,43-So1,44-OsSus1,45-Cc,46-Ca2,47-Nt,48-St2,49-Sl,50-StSus2,51- Nl1,52-Nl2,53-Ps1,54-Cp,55-Eg,56-Fa,57-Pv,58-PaSusy,59-GmSusy,60-Ps2,61-VfSusy,62-MsSusy,63-Mt,64-Psp,65-Ptm,66- Cr,67-Bv,68-Dc,69-Gh2,70-CitSus1,71-Bo,72-Av1,73-Asp1,74-9NOST,75-Ns1,76-Np1,77-Lm,78-Se,79-Gv,80-Np2,81-Ns2,82- Asp2,83-Av2,84-Sa,85-At,86-Dp,87-Da,88-No,89-Net,90-Neu,91-Nm,92-Pma,93-Pmb,94-Tm. The corresponding protein identifiers for these abbreviations are given in Table1. (1B) Parsimony tree derived using PAUP, with 10000 bootstrap, showing the separation of the 3 sucrose synthase subfamilies. The protein identifiers for the abbreviations used are given in Table1. (1C) Sub-familyA gene tree reconciled with species tree for the monocot species represented in this subfamily,reconciliation was done using the software Notung.(3 duplications, 2 losses) (1D) Sub-familyB gene tree reconciled with species tree for the dicot species represented in this subfamily, reconciliation was carried out using the software Notung. (Duplications are shown by ‘D’, 5 duplications, 2 losses).
These include a sequence each from Pyrus pyrifolia and Pyrus communis besides two homologs from Craterostigma plantagineum. Within the dicot family B, there is ample evidence of duplication and divergence. This feature is exemplified by these pairs- Medicago truncatula and M. sativa, Phaseolus aureus and P.vulgaris; Coffea arabica and C.canephora; N.langsdorfii and N.tabacum. This feature is seen in the reconciled tree (Figure. 1D).
In Family C with 16 representative sequences, there are 4 homologs from A. thaliana (SUS2, SUS3, SUS5 and SUS6; SUS5 and SUS6 cluster separately), two from C.plantagineum, one each from B.vulgaris, S.tuberosum, C.arabica and C.unshui. Even here, duplication and divergence events are evident. Homologs from Oryza and Maize are also seen in Family C along with the other dicot sequences. The Oryza SUS sequence has between 55 - 65% identity with all the four Arabidopsis sequence representatives in this family. The ATSUS5 and ATSUS6 proteins cluster separately within this family, as has been reported earlier (Baud et al., 2004).
Most sequences in sub-family A cluster with their closest plant relative. This family is represented by two wheat homologs, with 77% sequence identity (Wheat Q42223 and Wheat 082073); Maize Sus1 and Sus2 with 80% identity SUS1 and SUS2 sequences from Tulipa gesneriana with 85% identity; SUS1 and SUS2 sequences from H.vulgare with 78% sequence identity. Sub-family A also has two homologs from B.oldhamii with 81% identity; that these sequences arise out of duplication events is shown in Figure 1c. The Oryza sativa SUS2 and SUS3 proteins are nearly 90% identical. There are two homologs from P.distinctus that are both represented in Family B. Other Family B representative sequences include M. acuminata,O. goldiana and Lolium perenne. The sequences represented in subfamily A differ from their homologs in subfamilies B and C in the presence of two leucine zipper regions in many representatives of this family (LNDRIRSLDALQAALRKAEEHL, and LSAHTNELVAVFSRLNQGKGML) (Figure 2). Three monocot sequences in Family C have a variant of one of the bzip motifs (LTDNKKPLLDYLLALSHRGDKL in O. sativa and Maize species). This finding is consistent with reports on the role of a helix loop helix transcription factor in the regulation of carbohydrate metabolism in response to sugar levels in the human liver, and in the sucrose induced repression of translation (SIRT) response in plants (Weise et al., 2004). Sub-family B sequences are mostly characterized by four motifs for Asn-glycosylation (NYSD/ NHTD), protein kinase C and casein kinase 2 phosphorylation sites and the MYRISTL pattern. The presence of additional cAMP phosporylation site motif (KKLS/KRLT/ RRLT), ATP-GTP-A or leucine zipper motifs characterize the difference within the sub-groups in sub-family B representatives. Most Family C representatives have an amidation motif (GGKK/NGRR/QGKR) that is absent in the other two families. Features shared amongst all representatives of the three sub-families include Asn- glycosylation motifs (NYSD), PKC phosphorylation site patterns (SLR, TEK, SAK in subfamily B, SSK and TPK in Family A), casein kinase 2 phosphorylation patterns(SRFD, TYQE in sub-family C and TFQE in sub-families A and B) and MYRISTL (GQYESH, GVTQCT, GTEHTH, GNLVAS in B and C, with GLVAC/T in subfamily A). We note a feature of the MYRISTL motif (N-myristoylation site) in that some motifs are dicot sequence-specific (GILQS/HH, GL/ YPDTG) and differ between representatives of subfamilies B and C in a single amino acid. N-myristoylation is a lipid modification ensuring the proper function and intracellular trafficking of proteins involved in many signaling pathways. The presence of this feature suggests multiple roles of this enzyme and multiple localization of the protein helping them reside in more than one compartment of the cell. Attachment of the myristoyl residue provides hydrophobicity that influences the partitioning of proteins to cellular membranes and can serve to promote protein-protein interactions (Johnson et al., 1994). It has been shown that phosphorylation of sucrose synthase decreases its hydrophobicity and releases the protein from the membrane while dephosphorylation increases association with membranes (Winter et al., 1998). The existence of multiple phosphorylation, N-glycosylation and leucine zipper motifs has been reported in Maize SUS3 (Carlson et al., 2002). From this prediction, it appears that all plant sucrose synthase sequences have some or all of these patterns with variants that could be phylogenetic cluster specific. The sequence variations in sucrose synthase proteins were then examined from a structural perspective. A representative alignment of the sequences derived from all three subfamilies was further examined using ConSurf (http://consurf.tau.ac.il). This structure based sequence conservation analysis was made using the sucrose phosphate synthase (SPS, PDB 2r60) as a template. This is shown in Fig.3. As is evident from this figure, the variable regions are located in the N terminal region (7-8, 26-27 and 68-73) and a few sectors in the C terminus (398-401, 423-427, 447-448). The most variable segments comprise those between 239-253, 258-262 and 306-309. Although the functional significance of these variations is unclear (apart from a generalized assumption that these regions are not essential for function), it is possible that some of these segments might modulate the activities of these enzymes (allostery) or might be involved in interaction with other co-factors in the cellular mileau.

Figure 2: Sub-family A sequence representatives in Multiple sequence alignment (MSA) with sequence features highlighted and secondary structure shown under the alignment. The MSA was generated using ClustalW and edited in Jalview. Secondary structure was generated using JNetPred.
Presence of Target Peptides
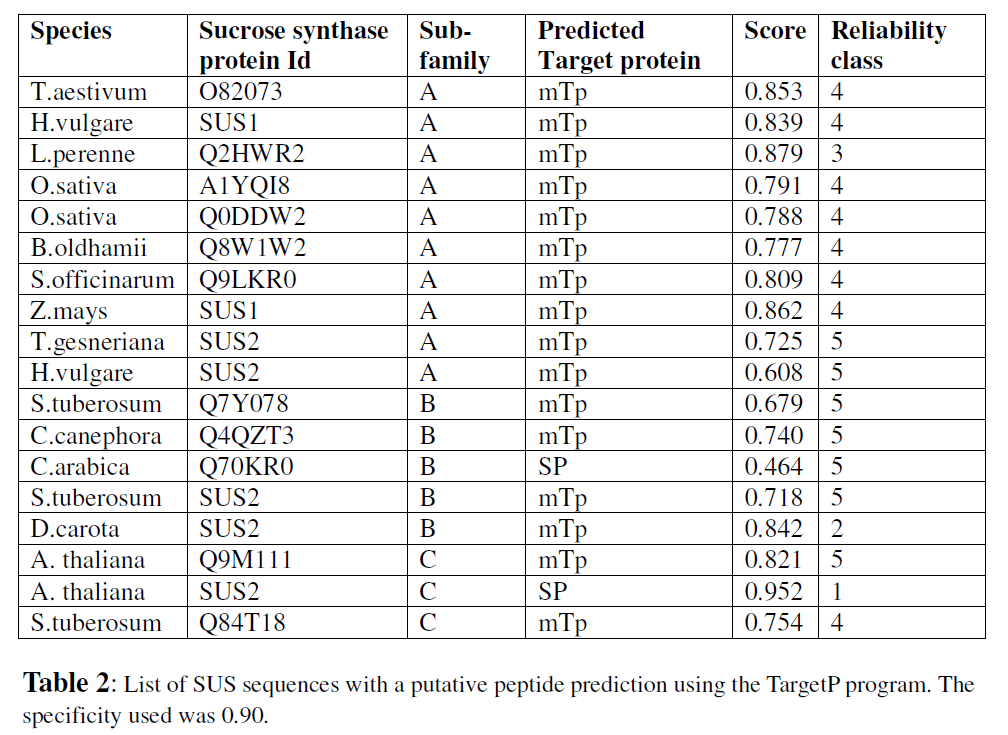
It has been suggested that the distribution of SUS in organelles not involved in sucrose metabolism may indicate functional roles beyond sucrose degradation (Gerrits et al., 2001; Oswald et al., 2001; Subbaiah et al., 2006). A related finding is that many sucrose sythase sequences show a mitochondrial targeting peptide (mTP). This study was carried out on maize sucrose synthase proteins (Subbaiah et al., 2006). Many of these polypeptides are synthesized in the cytosol and subsequently translocated to other organelles by virtue of a targeting sequence. The TargetP server was used to scan for these signal sequences. Based on the location of these target peptides at the N terminus of these proteins, it is seen that many more of sub-family A monocot representatives have a mitochondrial localization relative to subfamily B or C sequences.
Only 3 (8.5%) of subfamily B and 2 (12.5%) of subfamily C sequences were predicted to have the mTP peptide relative to 12 (63%) of subfamily A sequences (Table 2). The scores for each target peptide sequence were generated with specificity of>90. This sequence analysis suggests that one protein each from subfamilies B and C is likely to be involved in secretory pathways. While not much is known on the role of sucrose synthase in the mitochondria, it is suggested that its role may be similar to that of hexokinase interaction with mitochondrial VDAC (voltage dependent anion channel). This interaction regulates the opening of the mitochondrial permeability transition pore and also affects nutrient or sugar signaling and apoptosis (Azoulay-Zohar, 2003). The analysis of the SUS proteins in this dataset suggests that the presence of a targeting peptide is more of a monocot SUS sequence specific feature and may not apply across all plant sucrose synthase proteins.
Transmembrane Segments and Secondary Structure
The multiple sequence alignment of the protein sequences was carried out using the consistency based method of PROMALS (PROfile Multiple Alignment with predicted Local Structure . Sequence alignment with PROMALS is a two step process. In the first step, highly similar sequences are progressively aligned with a weighted sum-of-pairs measure of BLOSUM62 scores resulting in a set of sequences or pre-aligned groups that are relatively divergent from each other. In the second alignment stage, one representative sequence is selected from each pre-aligned group, and PSI-BLAST is used to search for homologs from the sequence database UNIREF90. The profiles generated here are derived from the PSI-BLAST alignments and PSIPRED secondary structure prediction. A matrix of posterior probabilities of matches between positions is obtained by forward and backward algorithms of a profile-profile hidden Markov model. These matrices are used to calculate a probabilistic consistency score and the representative sequences are further aligned according to a consistencybased scoring function. The pre-aligned groups obtained in the first stage are subsequently merged with the multiple alignments of the representatives. The accuracy of this protocol derives from the combined use of homologs from a structure database and secondary structure predictions using PSIPRED. While several available tools were used for MSA generation, the representative sequences derived from the PROMALS output were used to search the stress sequence database for homologs. The secondary structure predictions of sequences from the three families show minor differences in the N terminal regions containing the putative target peptide region (Fig.4).
While sucrose metabolism proteins are generally believed to be cytosolic proteins, predicted trans-membrane segments were seen in programs such as AveHas as well as TMpred (http://www.ch.embnet.org/software/TMPRED_form.html). Figure 4 shows the results on the amphipathicity, hydrophobicity and similarity of sequences in each of the three sub families generated using the AveHas programme. Sub family A is characterized by the presence of 11 amphipathic peaks (A1-A11) with the angle set at 180° (as is appropriate for aß-strand) and 6 hydropathic peaks (H1-H6). Sub family B is characterized by 9 amphipathic peaks (A1-A9) and 7 hydropathic peaks (H1-H7). Sub family C is characterized by 8 amphipathic peaks (A1-A8) and 7 hydropathic peaks (H1-H7). In all these three plots, the average similarity plot follows the hydrophobicity plot and the major difference between the three families is in the N-terminal region specifically between residues 7-90 in the alignment. We also note differences in the hydrophobic character of this region between families, with increased hydrophobicity in sub-family C while sequences from sub family A (monocots) are more amphipathic in nature. The most conserved region in the alignment of sub-family A representatives (150 – 445 residues) is characterized by fewer hydrophobic amino acid stretches (A5-A10 and H2-H4). The most conserved region of the alignment in sub family B (291-443) corresponds to H2-H5 and there is complete conservation in hydrophobic residues within this region in all the representatives. Predicted trans membrane helices also lie within this region (249-480). In sub family C, there is likelihood of trans membrane segments between position 20-80 of the aligned sequence besides one between the 270-295 positions of this alignment. Thus, within each of sucrose synthase family clusters, there appear to be structural differences that are not understood, but potentially contribute to the phylogenetic separation of the families and potential differences in their functional role in abiotic stress.

Figure 3: A representative multiple sequence alignment was mapped using CONSURF over the recently available three dimensional structure of the SPS enzyme (PDB:2r60). The coloring strategy is that followed by CONSURF, highly conserved residues are colored maroon, while highly variable residues are colored blue, with average residues in white.

Figure 4: Average hydrophobicity, amphipathicity and similarity plots for the multiple sequence alignment of sequences representing the 3 sucrose synthase subfamilies A, B and C. MSA was derived using ClustalW and plots were derived using the AveHas program.Alignment position is indicated at the bottom of the figures. A1-A10 indicate amphipathicity peaks, H1-H7 indicate hydrophobicity peaks.
Analysis of the Stress EST Database
The sequences identified using PROMALS represent sequences across all 3 subfamilies. There were 6 representatives from sub-family A, 8 from sub-family B and 11 from sub-family C. These sequences were searched against the EST databases for nine crops, five monocots (barley, sorghum, rice, maize and wheat) and 4 dicot species (Arabidopsis, Medicago, Soybean and Tomato). The reference sequences were also searched against the EST datasets derived from stress cDNA libraries from each of the nine crops. The percentage of identified hits to reference sequences from the three subfamilies in the EST database (not derived from abiotic stress) ranged from 0.008% (AtSus6, sub-family C against Rice EST database accessed at JCVI) to 0.04% (Eucalyptus grandis, sub-family B against the Maize database accessed at JCVI) with an average of 0.026%. The percentage of identified hits to reference sequences from the three families in the stress EST datasets for the nine crops ranges from 0.005% (all reference sequences against the Arabidopsis ESTs derived from stress cDNA libraries) to 0.44% (all reference sequence against the Tomato EST dataset derived from stress cDNA libraries) with an average of 0.08%. On the whole, 24 reference sequences when searched against stress cDNA libraries returned a larger number of hits than when searched against normal EST datasets that do not include sequences derived from stress libraries, after normalizing the number of hits for the size of datasets. The only exception was the Arabidopsis stress EST dataset with very little representation of sucrose synthase homologs. There was no significant difference between the number of homologs to reference sequences having both domains (sucrose synthase and glycosyl tranferase1) as compared to a reference sequence with only one of the two domains. Interestingly, homologs to AtSus6, OsA and DaucaSus2 were found to have a significantly greater representation than the other reference sequences in the stress derived EST datasets (5% significance using the Z-test) across six of the nine crops for which stress EST datasets were screened. The exceptions were Arabidopsis, Maize and Rice datasets where the percentage hits were higher in non-stress than stress datasets. AtSus6 and OsA belong to sub-family C and, in terms of sequence features, they are characterized by the bzip + amidation patterns, have transmembrane predictions and lack a target peptide region.
Dauca Sus2 from sub-family B has a mitochondrial target peptide but lacks the bzip and amidation pattern. In the Arabidopsis stress EST data collection, the paucity of transcripts to sucrose synthase could be because of the tissues from which the cDNA libraries were derived. While the high representation could well be the result of high transcript levels in certain libraries derived from particular tissues, many libraries are normalized whereby redundant transcripts from genes with high expression have been eliminated. The presence of a large number of sucrose synthase transcripts in datasets derived from stress cDNA libraries derived from the four abiotic conditions (heat, cold, drought and salt) is not new. High levels of AtSus1 mRNA has been known to be induced by cold (Baud et al., 2004; Dejardin et al., 1999; Martin et al., 1993) and drought (Baud et al., 2004; Dejardin et al., 1999; Pelah et al., 1997). AtSus2 transcripts are reported to increase preferentially due to oxygen deficiency (Dejardin et al., 1999). AtSus5 and AtSus6 are not known to respond to stress (Baud et al., 2004). This is relevant as the current analysis shows that AtSus6 is significantly over-represented in datasets derived from stress.
To summarize, sequences of sucrose synthase were examined from cyanobacteria, proteobacteria and plants. Plant sequences could be grouped into at least three distinct subfamilies. Sequences derived from dicotyledonous plants were distributed across two subfamilies whereas monocot sequences cluster into a single sub-family. These sub-families differ in their amino acid content, the presence of target peptides, protein length variations and secondary structural content. The presence of a target peptide suggesting mitochondrial localization is higher in sub-family A as compared to the other two sub-families. Mapping of the sequence features on a three dimensional SPS structural template shows distinct patterns of sequence conservation amongst sucrose metabolism enzymes. In terms of representation in stress derived EST datasets, there is a significant difference in expression of sucrose synthase sequences in stress derived EST datasets as compared to normal EST datasets. It would be interesting to examine the orthologous sequences derived in response to stress with their non-stressed counterparts for amino acid replacements and potential structural changes. The analysis of the evolutionary characteristics of this enzyme presents an important source of information with applications in agricultural biotechnology especially when transgenic plants are generated to asses the impact of sucrose synthase transgene expression on yield and quality parameters in commercially important tuber/fruit yielding angiosperms (Smith, 1999). This bioinformatics study could thus form a robust starting point to collate experimental evidence for the physiological significance, tissue specificity and functional significance of the variations in the protein sequences corresponding to the phylogenetic analysis reported here. Analysis such as these would be of particular relevance to biotechnology projects aiming to produce crops resistant to extreme environments.