Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Research Article - (2015) Volume 4, Issue 2
It is over seven decades since the seminal work of the otolaryngologist, Dr. Danely Slaughter was published. In patients with oral cancer, he critically demonstrated that carcinogen exposure led to the development of “condemned mucosa”, within which multiple cancers emanated. Thus, the histologically normal cells adjacent to the primary tumor harbor invisible features of the cancer. Other workers such as Strong have confirmed Slaughter’s initial observations, and in recent times by many investigators using molecular genetic approaches. This concept of carcinogenesis is partly responsible for our failure to successfully treat some of our cancer patients. With current technological advancements, biomarkers of field cancerization can be assayed using even noninvasive samples such as body fluids such as saliva and bronchial washes. In clinical practice, such validated biomarkers should enable risk stratification, and hence close surveillance of individuals at risk for early cancer detection or the early deployment of chemopreventive strategies. Arguably, this will immensely contribute to the reduction in the socioeconomic, physical and psychological burden of cancer.
<Keywords: Field Cancerization; Field Defect; Field of Injury; Field Effect; Condemned Mucosa; Damaged Mucosa; Cancer; Prevention; Early Detection; Chemoprevention
The tremendous achievements made over the past several decades in cancer research has contributed immensely to our ability to better detect and manage cancer today. However, the ongoing failures in our fight against most cancers are well known to clinicians, oncologists, palliative caregivers, patients and their families and friends including most of us. The traumatic experiences encountered due to cancer diagnosis and outcomes need to be minimized, if not completely eradicated, and this has been the goal of all individuals involved in cancer care and research. The unacceptable global burden of cancer requires multidisciplinary and paradigm-shifting approaches for curtailment. The estimated cancer incidence and mortality for the next two decades by Thun and colleagues [1] is very alarming and unacceptable. Their forecast estimates that by 2030, ~26 million new cancer cases will be diagnosed, with 17 million cancer deaths per year. It is also suggested that the distribution, types and possible etiologic agents of cancer will change with time. In the less developed parts of the world, it is projected there will be a 10% change in cancer incidences between 1975 (recorded incidence of 51%) and 2050 (estimated incidence of 61%). Even more worrisome is the emerging evidence that less developed nations will bear much of the cancer burden in the future. Indeed, the World Health Organization identifies a similar pattern, suggesting the need to heighten our efforts against the battle against cancer. While developed nations will show a modest decline in incidence and death rates, the reverse is true in the developing world, where deaths from cancer is expected to outpace deaths from HIV/AIDS, tuberculosis and malaria combined. While the picture in the developed world looks slightly different, there is no obvious evidence of any bright light at the end of the tunnel.
Contributing risk factors to this global cancer trend are well known. Lifestyle changes from our ancestors, such as our dependence on Western diet, tobacco and alcohol abuse, industrialization with its associated chemical exposures, as well as physical inactivity are all risk factors for the development of various cancers. Moreover, our ineffective treatment of infectious diseases, such as Helicobacter pylorigastritis and other carcinogenic viruses contributes to the elevated cancer cases in some geographic regions. All these factors are causative agents of precancer fields or “field cancerization” in tissues and organs. Obviously, positive lifestyle alterations appear to be one of the most important preventable interventions that could help reverse this global trend. However, because habits are often difficult to change, complementary deployment of other strategies such as targeting genetic footprints of carcinogens has potential for early detection, chemoprevention, diagnosis, and effective cancer management. Thus, to reverse the emerging scenario, cancer, I believe, must be tackled at its “genesis and not exodus”. The term “field cancerization”, introduced by Slaughter and colleagues, is also referred to as condemned mucosa with reference to oral epithelium, and in current terms as field effect, field defect, and field of injury. Common to all these terminologies is the “damaged-field” concept of cancer development. Slaughter first recognized this phenomenon in his review of the origin of multiple tumors in an individual, at about the era of the Second World War [2]. However, he and his colleagues introduced the phrase “field cancerization” later in 1953 [3] when they stated: “From the foregoing observations it would appear that epidermoid carcinoma of the oral stratified squamous epithelium originates by a process of “field cancerization,” in which an area of epithelium has been preconditioned by an as-yet-unknown carcinogenic agent”. Slaughter et al., 1953 The observations of these early scientists were that cancer did not develop in a focal fashion amenable to simple complete surgical resection. Slaughter and his contemporaries were well convinced by their observations that a large expanse of tissue was affected and hence at risk of giving rise not only to one, but multiple cancers. The description by D. P. Slaughter [2,3], and a decade later by M. S. Strong [4] indicate global damage to oral epithelium and contiguous upper aerodigestive structures by carcinogens (tobacco and alcohol), leading to the evolution of multiple primary tumors. This initial concept is well articulated in Slaughter’s 1944 statement: “From the foregoing presentation it seems apparent that cancer does not arise as an isolated cellular phenomenon, but rather as an anaplastic tendency involving many cells at once. The studies of “carcinoma in situ” demonstrate this for many epithelial areas, but of course it does not necessarily follow that all malignant tumors arise in this fashion”. Slaughter, 1944 It is evident from this statement that Slaughter also recognized the possibility of other mechanisms of carcinogenesis. Current cell biology and molecular genetic studies suggest a much broader definition of field cancerization. This concept has a much deeper connotation that cannot be limited to only contiguous structures exposed to carcinogens, but noncontiguous tissues such as single organs or portions of organs (e.g., prostate), paired organs (e.g., breasts), as well as organs of the same developmental origins. Moreover, non-epithelial structures such as the hematologic and nervous systems can have “field” changes as well [5]. Conceivably, the whole body can be predisposed to cancer development as a result of genetic defects inherited or acquired during embryogenesis (e.g., Li-Fraumeni syndrome). Clearly, Slaughter was cognizant of this fact when he wrote: “On the other hand, the multiple tumors that occur in different organs suggest a tumor diathesis in the individual, certainly a greater degree of susceptibility than can be explained away as mere coincidence or accident”. Slaughter, 1944 Obviously, the field effect phenomenon of cancer is now well appreciated to involve more than just a few carcinogenic agents. A current and all-inclusive perspective of field cancerization should broaden the causative agents and factors to include a diverse range of both exogenous and endogenous factors. The exogenous agents include chemical carcinogens such as in tobacco smoke, physical agents such as ultraviolet radiation, and infectious agents such as Helicobacter pylori, human papilloma and hepatitis B viruses. Endocrine secretions (e.g., estrogens, testosterone), exocrine secretions (e.g., gastric acid), and even inherited cancer mutations (e.g., BRCA 1, 2) constitute the endogenous causative factors of cancer fields. Whereas the field effect phenomenon is well known by scientists and oncologists, the exact mechanism of cancer field development is unclear. There are likely to be multiple mechanisms that operate differently in various organs and tissues. For instance, in an epithelium, a carcinogen could induce diverse or similar genetic alterations in many cells leading to polyclonal identical or divergent lesions poised for cancer evolution (Figure 1, a model of prostate cancer field cancerization). This will be reminiscent of Slaughter’s observations of an expanse epithelial preconditioning. Similarly, in contiguous epithelium, the possibility of a single clonal expansion could potentially create a cancer field. Expansion of inherited mutations, or errors that occur early during embryogenesis could create global preconditioned structures for cancer development in the future. What is also well demonstrated in several studies is the communication between cancer cells and the histologically normal adjacent cells, through epithelial–stromal signaling [6]. The established fact is that cancer is a genetic disease initiated often by epigenetic alterations, somatic mutations induced by carcinogens, as well as heritable mutations in cancer susceptibility genes. Cancer, however, is unlike the common cold that suddenly strikes. It takes several decades for a cell or group of cells to accumulate enough genetic alterations to become transformed into histologically overt malignant cells. Indeed, cancer cells evolve through hundreds of generations, acquiring enabling mutations over decades. This wide window of carcinogenesis offers enormous potential for primary prevention, early detection and diagnosis, chemoprevention, and surveillance. The concept of field changes offers opportunity for the discovery, validation and standardization of biomarkers for cancer management, including prognostication. Apart from the clinical applications, our knowledge of cancer initiation, and importantly how multiple synchronous and metachronous tumors develop are broadened by this concept. Tumor recurrences, whether second primary tumors or second field tumors are well-engrained in the minds of many current surgical and radiation oncologists, just as was appreciated and documented by D. P. Slaughter [3] and M. S. Strong [4] several decades ago: “Since surgically we are seldom beyond the limits of abnormal epithelium and since radiation notoriously is less effective on benign epithelium and leukoplakia than on carcinoma cells, local recurrence may be due to benign epithelium preconditioned to change toward cancer, which has been opposed in a suture line after excision of a tumor or has healed over the site of a tumor destroyed by radiation”. Slaughter et al., 1953 “Field cancerization should be taken into account in treatment planning of a patient with cancer so that all treatment options, including the use of radiation therapy, be kept open for as long as possible in the event that the patient may develop multiple primary tumors”. Strong et al., 1984.
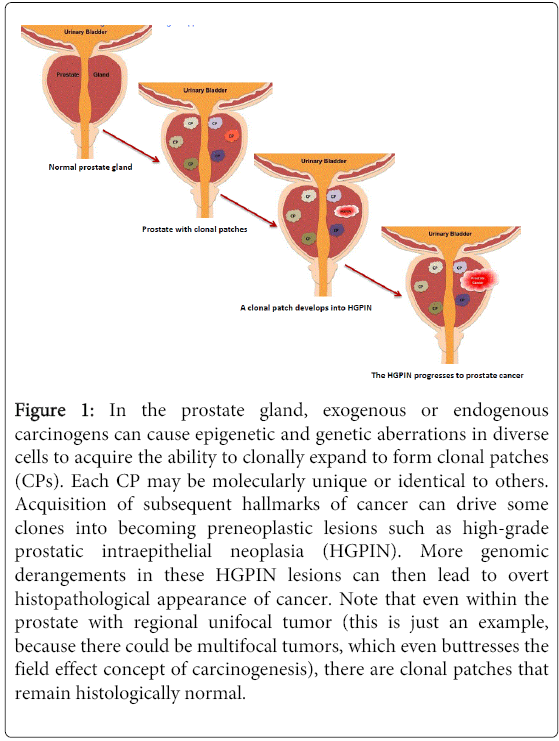
Figure 1: In the prostate gland, exogenous or endogenous carcinogens can cause epigenetic and genetic aberrations in diverse cells to acquire the ability to clonally expand to form clonal patches (CPs). Each CP may be molecularly unique or identical to others. Acquisition of subsequent hallmarks of cancer can drive some clones into becoming preneoplastic lesions such as high-grade prostatic intraepithelial neoplasia (HGPIN). More genomic derangements in these HGPIN lesions can then lead to overt histopathological appearance of cancer. Note that even within the prostate with regional unifocal tumor (this is just an example, because there could be multifocal tumors, which even buttresses the field effect concept of carcinogenesis), there are clonal patches that remain histologically normal.
Auerbach and colleagues elegantly demonstrated the tobacco-induced lung field defect in 1961 [29]. Their findings were consistent with tobacco-related histological changes in lung epithelia, with the development of widespread multifocal premalignant lesions. Because smoking is a known risk factor for development of lung cancer, studies have established a molecular field defect in smokers with or without cancer. In addition to other carcinogens, toxins from tobacco smoke are established agents that create the lung cancer field, either directly or through induction of epithelial inflammation. About 85% of smokers are at risk for developing lung cancer (that is having an established lung cancer fields evidenced by molecular alterations), but only ~15% will actually progress to develop the disease. Smoking causes aberrant promoter methylation of multiple genes in the epithelium of cancer-free individuals. These genes include cyclin-dependent kinase inhibitor 2A (CDKN2A), death-associated protein kinase (DAPK), glutathione S-transferase pi 1 (GSTP1), retinoic acid receptor β2 (RAR-β2), ras association domain-containing protein 1 (RASSF1A), cadherin 13 (CDH13), and adenomatous polyposis coli (APC). Thus, smokers have multiple foci of “damaged” epithelium with epigenetic alterations. Many lungs from smokers without cancer demonstrate numerous genetic changes as well, including loss of heterozygosity (LOH) and microsatellite alterations. Even in bronchial washes from both ipsilateral and contralateral lungs from smokers, LOH and tumor protein p53 (TP53) mutations are present. Additionally, in histologically normal lung tissue adjacent to lung cancer, mutations in epidermal growth factor receptor (EGFR), Kirsten rat sarcoma viral oncogene homolog (KRAS), LOH at chromosome 3p that harbors DDUT and fragile histidine triad (FHIT), and the loci of CDKN2A, chromosome 9p are characterized. Table 1 contain summary of the molecular alterations in smoke-associated pulmonary field cancerization, and references to subject matter alluded to in this section. While this is the tip of the iceberg for lung cancer, molecular field cancerization occurs in all tumors.
| Biomarker | Field Effect Changes | Reference |
|---|---|---|
| DAPK, RARb2, H-CADHERIN, CDKN2A, APC, GSTP1, RASSF1A | Promoter methylation | [7-11] |
| Microsatellite (MS) repeats | Increased MS repeats | [7,12,13] |
| Alleles | Allelic loss or LOH | [14,15] |
| Chromosome 3p: (FHIT, DDUT) Chromosome 9p: (CDKN2A) |
LOH | [7] |
| Genomic instability RNF20 and SSBP2 RASGRP3 |
Genomic instability Loss of RNF20 and SSBP2 Amplification of RASGRP3 |
[16] |
| TP53, EGFR, KRAS | Mutations | [17-20] |
| Global transcriptomics | Altered expression | [21,22] |
| Antioxidant gene signature | Upregulated 16 antioxidant genes | [23] |
| 80-gene signature | Altered expression | [24] |
| PI3K pathway activating genes | Increases expression of pathway genes | [25] |
| miRNA | Expressional changes | [26] |
| Global transcriptomics LAPTM4B |
Expressional changes LAPTM4B upregulation |
[27] |
| mRNA and miRNA expression ERK/MAPK pathway |
Expressional changes Altered ERK/MAPK pathway activation in contralateral lung |
[28] |
Table 1: Biomarkers of field cancerization in the lung
Over 70 years since Slaughter’s ingenious findings, there is currently overwhelming molecular pathologic evidence in support of his initial observations. Numerous epigenetic and genetic alterations as a consequence of carcinogen exposure drive a cell into uncontrollable proliferation. The onus is now upon us to harness these molecular imprints in cancer fields for the effective management of our patients. With advancing technological developments, including single cell genomics and deep sequencing, we should be able to profile body fluids in a noninvasive fashion for early risk biomarker detection, and hence the possibility to administer chemopreventive therapies. The importance of Slaughter’s field cancerization concept in surgical oncology, chemotherapy and radiotherapy is much appreciated today, but we still need more knowledge and validated biomarkers for clinical translation. The eventual adoption of such new paradigms in cancer management should contribute significantly to our fight to conquer cancer, and reduce the global psychological, physical and socio-economic burden of this disease.