Journal of Developing Drugs
Open Access
ISSN: 2329-6631
![]() +44 1478 350008
+44 1478 350008
ISSN: 2329-6631
![]() +44 1478 350008
+44 1478 350008
Research Article - (2017) Volume 6, Issue 3
In the present study, Gliclazide is a second-generation sulfonyl urea derivative used to treat non-insulin dependent diabetes mellitus. The drug has been classified as class-II drug according to biopharmaceutical classification system having low solubility and high permeability. So, an attempt was made to enhance the solubility of gliclazide by solvent evaporation technique. A significant enhancement of gliclazide dissolution rate will be obtained using by using suitable techniques. In this PVP-k30, polyethylene glycol and soluplus were mixed with the drug in different ratios (1:1, 1:3) and prepare P1, P2, PE1, PE2, S1 and S2 solid dispersion batches. The optimized solid dispersions P1 were further kneaded with suitable proportions of super disintegrants such as; Cross carmellose and Sodium starch glycolate. Dissolution profile will show decrease amount of drug release at specified time. DSC as well as X-ray diffraction showed reduced drug crystallinity in SDs. Scanning electron microscopy and particle size analysis revealed significant decreased particle size of the drug in SDs. FT-IR spectroscopy demonstrated no detectable interactions between the drug and excipients. On that the P1 batch will be consider for eight different fast dissolving tablets Preparation. The prepared FDT’s were evaluated for various parameters like disintegration time, wetting time, drug content, in vitro drug release study etc. and shows the satisfactory result. The formulation of F-4 containing cross carmellose sodium (5%) showed better result in disintegration time 11 sec. and maximum in vitro drug release of 99.89% at the end of 40 minutes.
<Keywords: Gliclazide; Fast dissolving tablet; Solid dispersion; Solvent evaporation method
Fast Dissolving Tablets (FDTs) are solid single-unit dosage forms that are placed in mouth, allowed to disperse/dissolve in the saliva without the need of water and provide a quick onset of action. Fast dissolving tablets are formulated by using super disintegrants like cross carmellose sodium, cross povidone and sodium starch glycolate. The various technologies used for manufacturing of FDTs are Freeze drying, Spray drying, Molding, Mass extrusion, Melt granulation, Sublimation and Direct compression.
The enhanced dissolution rate characteristics of solid dispersions can generally be accounted for one of the following mechanisms: eutectic formation, increased surface area of the drug due to precipitation in the carrier, solid solution formation, and improved wettability due to intimate contact with hydrophilic carrier, precipitation as a metastable crystalline form or a decrease in substance crystallinity. Both the properties of carrier-drug combination and the method of manufacturing will influence the type of solid dispersion formed and thus subsequent behavior of the solid dispersion [1].
The PVP, PEG and Soluplus are the most used drug carrier for solid dispersion preparation [2-4] due to their strong hydrophilic properties and their capability to form molecular adducts with many compounds. The presence of hydroxyl and carbonyl group in repeat units of these polymers tends to increase the water solubility [5-8] and stability [9,10] of the drug and also improve its bioavailability [11]. It is accepted nowadays that the glassy state of drug compound dispersed in such polymer matrices is the most desired state, since it improves its dissolution rate and hence drug absorption.
Gliclazide shows poor water solubility (55 μg/ml) and dissolution properties and this can give rise to low and erratic bioavailability and poor dose-effect proportionality. Moreover, gliclazide being a weak acid (pKa 5.8) its solubility is lower in acid medium and this could be a problem in the design of an immediate release oral dosage form because the entire dose should be promptly released in very short times in the upper part of the gastrointestinal tract. Therefore, the improvement of gliclazide dissolution behavior is an important issue for enhancing its bioavailability and therapeutic efficacy.
Materials
Gliclazide were obtained as a gift sample from Sun Pharma, Mumbai. Croscarmellose sodium, sodium starch glycolate and all other ingredients used were of laboratory grade.
Preparation of solid dispersion of gliclazide solvent evaporation method
Different formulations of solid dispersions of GLZ were prepared two ratios with each polymers PVP K-30, PEG 6000 and soluplus by solvent evaporation method. Accurately weighed quantities of drug and carrier were homogenously dissolved in sufficient volume of ethanol as a volatile solvent. The solution was allowed to evaporate at ambient temperature and the resulting solid mass was then dried in a desiccator. Solid dispersions were sieved through sieve number 60 to confirm the size uniformity (Table 1).
| Carrier | Batch | Ratios |
|---|---|---|
| PVP K-30 | P1 | 1:1 |
| P2 | 1:3 | |
| PEG 6000 | PE1 | 1:1 |
| PE2 | 1:3 | |
| Soluplus | S1 | 1:1 |
| S2 | 1:3 |
Table 1: Batches of solid dispersion prepare with drug: polymer ratio.
Evaluation of solid dispersion batches
Determination of drug content: The Solid dispersions equivalent to 10 mg of GLZ were weighed accurately and dissolved in 50 ml of ethanol and stirred well with magnetic stirrer. The drug content was determined at 217 nm by UV spectrophotometric analysis after suitable dilutions.
Determination of percentage yield: The percentage yields of gliclazide were determining using formula:

Where, P=practical yield (mg); T=theoretical yield (mg).
Solubility studies: Known equivalent amount of different solid dispersion batches were added to 25 ml distilled water in conical flask add bead into it. Then this conical flask was shaken using mechanical stirrer for 24 hours. After 24 hours sample was filter using watmann paper no.4. dilute suitably and analyzed spectrophotometrically at 217 nm.
Fourier transform infra-red spectroscopy: FTIR spectra of GLZ and solid dispersions were recorded using a FTIR Spectrophotometer (cary 630 FTIR spectroscopy, agilent technologies, united states). Samples were prepared using KBr (spectroscopic grade) disks by means of hydraulic pellet press at a pressure of 7-10 tons. The samples were scanned from 4000 to 400 cm–1 at resolution of 4 cm-1.
In vitro dissolution study for batches: The in vitro dissolution study was performed using USP Type-II paddle dissolution test apparatus (Electrolab TDT-06P, India). The samples equivalent to 80 mg GLZ were placed in dissolution vessel containing 900 ml phosphate buffer as a dissolution medium (pH 7.4) maintained at 37°C ± 0.5°C and stirred at 100 rpm. 5 ml aliquot samples were collected periodically and replaced with a fresh dissolution medium to maintain sink conditions. After filtration through 0.45 μm membrane filter, GLZ was estimated spectrophotometrically at 217 nm with suitable dilutions. Same procedure for solid dispesion batches. On the basis of dissolution study P1, PE1 and S1 are selected.
Thermal analysis: DSC thermograms were useful tool to assess the chemical compatibility as well as the degree of crystallinity of excipients and drugs. The thermal behavior of the GLZ: PVP K-30 (1:1), PEG 6000 (1:1) and soluplus (1:1) formulations were determined using a differential scanning calorimeter (pyris 6 DSC, Perkin Elmer Pvt. Ltd) for identifying the chemical compatibility of drug and polymers. Samples (3-5 mg) were accurately weighed into 50 mg aluminium pans and hermetically sealed. The samples were heated from 0 to 300°C at a rate of 10°C/min under a dry nitrogen gas purge.
X-Ray Diffraction (XRD): Degree of crystallinity is one of the factors influencing the solubility and dissolution rate. Hence, crystalline nature of substances and extent of its conversion to amorphous form was studied by X-Ray Diffractometry (XRD). XRD patterns were studied using X-Ray diffractometer (brukeraxs system, D8, Germany). Samples were irradiated with Cr- radiation of wavelength 2.289 A° and analyzed between room temperature (2θ).
Scanning Electron Microscopy (SEM): Morphology of GLZ and batches were examined under scanning electron microscope (JEOL JSM-6360A, Hitachi ltd., Japan) at an acceleration voltage of 10 kv. Sample were sputter coated (E-1010 Hitachi Ltd., Japan) with gold palladium and observe at different magnification.
Preparation of Fast Dissolving Tablet
The Fast dissolving tablets of Gliclazide were prepared by using super disintegrants- cross carmellose sodium and sodium starch glycolate at different concentrations alone and in combination by direct compression method as shown in Table 2. All material was accurately weighed. All the materials were passed through 60# screen prior to mixing and transferred to glass mortar and triturated till it was mixed uniformly. The mixture is then evaluated for precompression parameters. After this the powder mixture was compressed into tablets by using 9 mm diameter punch in a rotary tablet machine [12,13].
| S.No | Drug/Excipients | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Batch P1 (mg) | 80 | 80 | 80 | 80 | 80 | 80 | 80 | 80 |
| 2 | Cross carmellose sodium (mg) | -- | -- | 10 | 12.5 | 10 | 12.5 | 10 | 12.5 |
| 3 | SSG (mg) | 10 | 15 | -- | -- | 10 | 10 | 15 | 15 |
| 4 | MCC (mg) | 119.75 | 114.75 | 119.75 | 117.25 | 109.75 | 114.75 | 119.75 | 117.25 |
| 5 | Mannitol (mg) | 37.5 | 37.5 | 37.5 | 37.5 | 37.5 | 37.5 | 37.5 | 37.5 |
| 6 | Mg stearate (mg) | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 |
| 7 | Talc (mg) | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 | 0.625 |
| 8 | Sodium saccharin (mg) | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| TOTAL | 250 | 250 | 250 | 250 | 250 | 250 | 250 | 250 |
Table 2: Formula for preparing fast dissolving tablet of gliclazide.
Precompression Evaluation of Powder
Bulk density
Apparent bulk density (ρb) was determined by pouring blend into a graduated cylinder. The bulk volume (Vb) and weight of powder (M) was determined. The bulk density was calculated using the following formula:
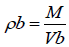
Tapped density
The measuring cylinder containing known mass of blend was tapped for a fixed time. The minimum volume (Vt) occupied in the cylinder and weight (M) of the blend as measured. The tapped density (ρt) was calculated using the following formula:
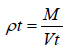
Angle of repose
Angle of repose was determined using fixed funnel method. The blend was poured through a funnel that can be raised vertically until a maximum cone height (h) was obtained. Radius of the heap (r) was measured and angle of repose was calculated using following formula:
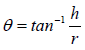
where, θ is angle of repose; h is height of pile; r is the radius of the base pile.
Hausner ratio
Hausner ratio is an in direct index of ease of powder flow. It is calculated by the following formula:
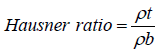
where ρt is tapped density; ρb is bulk density.
Lower Hausner ratio (< 1.25) indicate better flow properties.
Carr’s compressibility index
The simplex way of measurement of the free flow of powder is compressibility, an indication of the ease with which a material can be induced to flow is given by compressibility index of the granules was determined by Carr’s compressibility index (I) which is calculated by using the following formula:
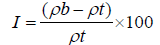
Evaluation of Fast Dissolving Tablet
Weight variation
The weight variation test is carried out in order to ensure uniformity in the weight of tablets in a batch. The total weight of 20 tablets from each formulation was determined and the average was calculated. The individual weights of the tablets were also determined accurately and the weight variation was calculated.
Hardness
The hardness of tablet is an indication of its strength. Measuring the force required to break the tablet across tests it. The force is measured in kg and the hardness of about 3-5 kg/cm2 is considered to be satisfactory for uncoated tablets. Hardness of 10 tablets from each formulation was determined by Monsanto hardness tester.
Friability test
Friability is the loss of weight of tablet in the container due to removal of fine particles from the surface. Friability test is carried out to access the ability of the tablet to withstand abrasion in packaging, handling and transport. Roche friabilator was employed for finding the friability of the tablets. 20 tablets from each formulation were weighed and placed in Roche friabilator that rotated at 25 rpm for 4 minutes. The tablets were dedusted and weighed again. The percentage of weight loss was calculated again. The percentage of weight loss was calculated using the formula:
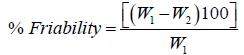
where, W1=Weight of tablet before test; W2=Weight of tablet after test.
Disintegration test
The USP device to rest disintegration was six glass tubes that are “3 long, open at the top, and held against 10” screen at the bottom end of the basket rack assembly. One tablet is placed in each tube and the basket rack is poisoned in 1 liter beaker of distilled water at 37 ± 2°C, such that the tablets remain below the surface of the liquid on their upward movement and descend not closer than 2.5 cm from the bottom of the beaker.
Uniformity of dispersion
Two tablets were kept in 100 ml water and gently stirred for 2 minutes. The dispersion was passed through 22 meshes. The tablets were considered to pass the test if no residue remained on the screen.
Wetting time
The wetting time of the tablets was measured using a simple procedure. Five circular tissue papers of 10 cm diameter were placed in a petridish containing 0.2% w/v methylene blue solution (3 ml). A tablet was carefully placed on the surface of the tissue paper. The time required for develop blue color on the upper surface of the tablets was noted as the wetting time.
Water absorption ratio
A small piece of tissue paper folded twice was placed in a small petridish containing 6 ml of water. A tablet was put on the paper and the time required for complete wetting was measured. The wetted tablet was then reweighed. Water absorption ratio, R was determined by using following formula were given:
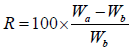
Wb is the weight of tablet before water absorption; Wa is the weight of tablet after water absorption.
In vitro drug release studies
The Gliclazide fast dissolving tablets were subjected to in vitro drug release studies in pH 7.4 phosphate buffer for 30 minutes to access the ability of the formulation for providing immediate drug delivery [14]. Drug release studies were carried out in eight stage dissolution test apparatus (DISSO 2000, Lab India) using 900 ml of dissolution medium (pH 7.4 phosphate buffer) maintained at 37 ± 10°C. The tablets were kept in the cylindrical basket and rotated at 100 rpm.
A successful attempt was made to formulate and evaluate fast dissolving tablet of gliclazide using solvent evaporation method. The Preformulation study was carried out for GLZ in the form organoleptic properties, melting point determination, and saturated solubility analysis, determination of λmax and calibration curve. The GLZ solid dispersion (solvent evaporation method) was prepared with polymers (PVP K-30, PEG 6000, and soluplus) in different proportion. The optimized P1 batches where used in preparation of fast dissolving tablet of gliclazide.
Preformulation studies
Characterization and identification of drug: Pure drug GLZ was characterized and identified by organoleptic properties, physical nature melting point and solubility (Table 3).
| S. No | Tests | Observation |
|---|---|---|
| 1 | Description | A white powder |
| 2 | Taste | Tasteless |
| 3 | Nature | Crystalline |
| 4 | Melting point | 180-182°C |
| 5 | Solubility | Insoluble in water, soluble in methylene chloride, acetone and alcohol. |
Table 3: Characterization for identification of drug.
UV spectroscopy analysis:
Determination of λmax : For characterization of drug by UV spectroscopy, it was important to know wavelength of maximum absorption (λmax ). The spectrum of gliclazide in ethanol was determined it shown in Figure 1. The wavelength of maximum absorption (λmax ) in different solvent such as water, ethanol, phosphate buffer and methanol were observed and it shown in Table 4; λmax of GLZ were found to be 217 nm in ethanol. The wavelength of maximum absorbance was considered for further A. Determination of λmax studies.
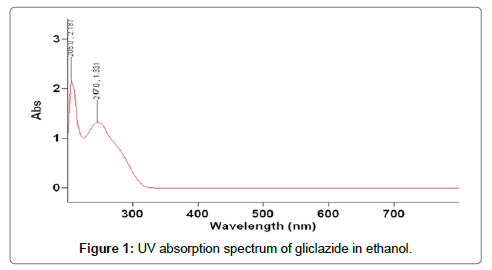
Figure 1: UV absorption spectrum of gliclazide in ethanol.
| S. No | Solvent | λmax (nm) |
|---|---|---|
| 1 | Ethanol | 217 |
| 2 | Water | 218 |
| 3 | Phosphate buffer 7.4 | 219 |
Table 4: Wavelength of maximum absorption (λmax) in different solvents.
Determination of calibration curves: The graph of absorption vs. concentration for pure gliclazide was found to be linear in concentration range 0-5 μg/ml at wavelength of maximum absorption shown in Table 5. Calibration curves and values of slope, intercept and R2 in ethanol, water and phosphate buffer 7.4 are shown in Figures 2-4 respectively.
| S. No | Solvent | Slope | Intercept | R2 |
|---|---|---|---|---|
| 1 | Ethanol | 0.068 | 0.0095 | 0.999 |
| 2 | water | 0.186 | 0.0016 | 0.999 |
| 3 | Phosphate buffer 7.4 | 0.075 | 0.024 | 0.999 |
Table 5: Calibration curve values of gliclazide in different solvent.
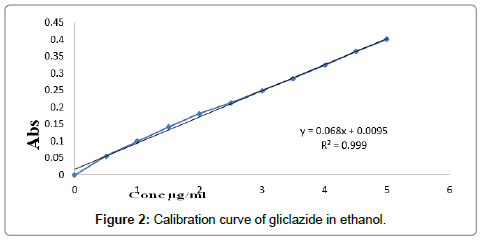
Figure 2: Calibration curve of gliclazide in ethanol.
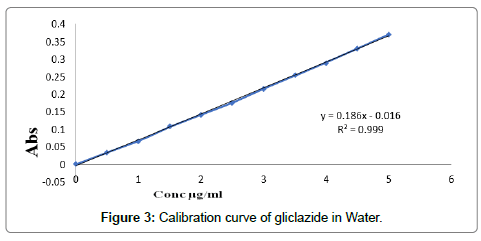
Figure 3: Calibration curve of gliclazide in Water.
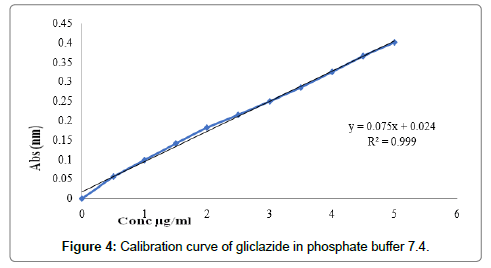
Figure 4: Calibration curve of gliclazide in phosphate buffer 7.4.
Saturated solubility of gliclazide and solid dispersion batches: Saturated solubility is important parameter that will affect the bioavailability of drug because of its poor solubility in aqueous media it possesses limitation in absorption of drug. Here saturation solubility of gliclazide was performed in water [15].
Saturation solubility of pure gliclazide and solid dispersion batches in water was shown in table from the result it suggested that gliclazide has very less solubility in water (Figure 5).
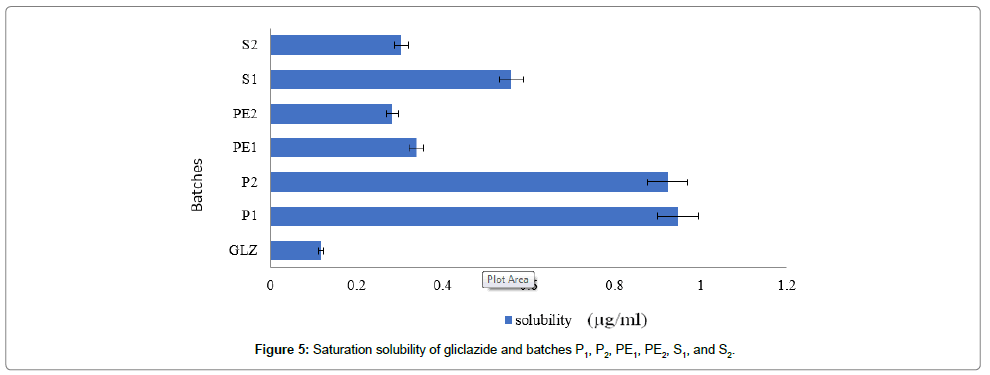
Figure 5: Saturation solubility of gliclazide and batches P1, P2, PE1, PE2, S1, and S2.
The all batches show higher saturation solubility than GLZ. As compared solid dispersion batches P1 and P2 Show higher than other batches because PVP is a synthetic water-soluble linear polymer; its inclusion in dry-form pharmaceuticals enhances the release and bioavailability of drugs of low solubility. PEGs have good solubility in water and have melting points below 65°C. These increases in solubility enhancement can be attributed due to formation of amorphous form of drug [16,17]. The Saturation solubility of gliclazide and solid dispersion batches P1, P2, PE1, PE2, S1 and S2 in water shown in Table 6.
| S. No | Batches | Saturation solubility (µg/ml) |
|---|---|---|
| 1 | Gliclazide (GLZ) | 0.1172 ± 0.009 |
| 2 | P1 | 0.9473 ± 0.007 |
| 3 | P2 | 0.9231 ± 0.01 |
| 4 | PE1 | 0.3384 ± 0.009 |
| 5 | PE2 | 0.2837 ± 0.006 |
| 6 | S1 | 0.5598 ± 0.007 |
| 7 | S2 | 0.3039 ± 0.01 |
Table 6: Saturation solubility of gliclazide and solid dispersion batches.
Drug content and percentage yield of batches: Drug content was determining using UV spectroscopy and percentage yield calculated by using previously mention formula. The results shown in Table 7. The drug content was determined at 217 nm by UV spectrophotometric analysis after suitable dilutions. The drug content of S2 was higher than other batches because soluplus having stuck in nature [18]. Other in case we select P1, PE2 and S2 batches prepare at different polymer on the basis of the drug content because it is showing higher drug content with comparative batches. On the basis of percentage yield P1, PE1 and S1 we select it.
| S. No | Batches | Drug content (%) | Percentage yield (%) |
|---|---|---|---|
| 1 | P1 | 69.14 | 35.15 |
| 2 | P2 | 60.6 | 30.23 |
| 3 | PE1 | 75.78 | 33.45 |
| 4 | PE2 | 82.68 | 23.09 |
| 5 | S1 | 67.38 | 23.08 |
| 6 | S2 | 88.54 | 16.07 |
Table 7: Drug content and percentage yield of batches.
In vitro dissolution study for batches: The in vitro dissolution profile of gliclazide and batches in phosphate buffer 7.4. In case of dissolution of gliclazide 51.75% drug release was observed in 10 min. In case drug and PVP K-30% drug release of P1 and P2 shows 75.71% and 49.27% respectively in 10 min. PVP acts as surface acting agents and facilities wetting of by decreasing the interfacial tension with dissolution medium. Drug release of PE1, PE2, S1 and S2 was found to be 67.61%, 51.63%, 68.96% and 53.21% respectively. On the basis of dissolution study, we select P1, PE1 and S1 select for further study because of higher the drug release at low time have high solubility of batch [19]. The S2 shows stick in nature as compared to S1 then it shows low dissolution rate. P1 and PE1 powder shows free flowing properties then it increases higher dissolution rate (Figure 6).
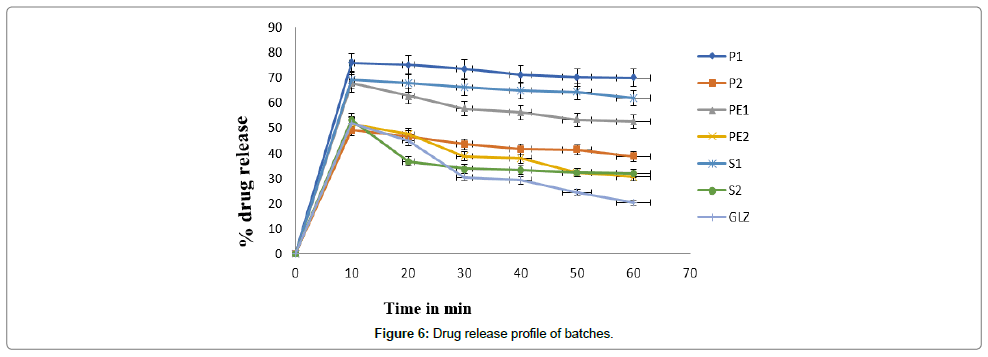
Figure 6: Drug release profile of batches.
Fourier transform infra-red spectroscopy: The gliclazide spectrum exhibit characteristics peak was shown in Table 8. The FTIR spectrum of GLZ and Batch P1 spectra were gave peak intensity for the free carbonyl group stretching mode at 1706 cm-1 and 1709 cm-1 respectively. For C-H stretching vibration of GLZ show the characteristic peak at 2935 cm-1 that slightly shifted in batch P1, P2, PE1, PE2, S1 and S2 at 2837, 2957, 2881, 2881, 2936, 2930 cm-1 respectively. The FTIR spectrum of GLZ obtained characteristic peak was slightly shifted in batches spectra. P2, PE2 and S2 shows boarden peaks as compare to other batches. These results indicated that GLZ and P1, PE1 and S1 interact via strong hydrogen bonds then shifting formulation from crystalline to amorphous. This indicated the no difference in the internal structures and conformation of these samples and the no interaction between the drug and the polymer (Figures 7-13).
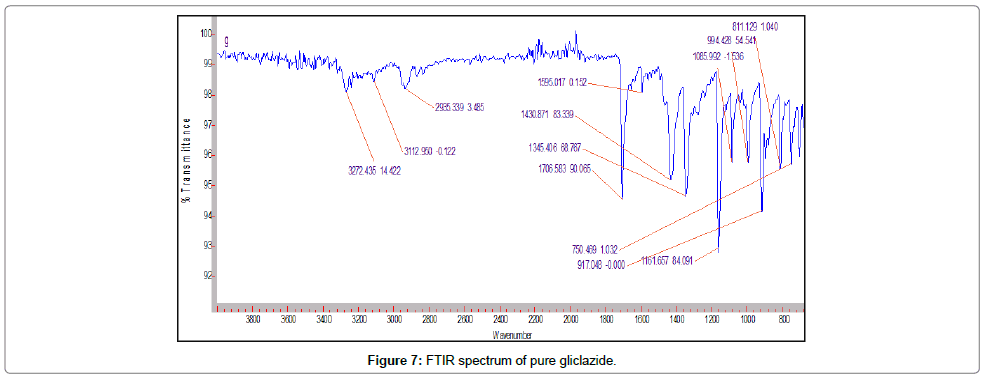
Figure 7: FTIR spectrum of pure gliclazide.
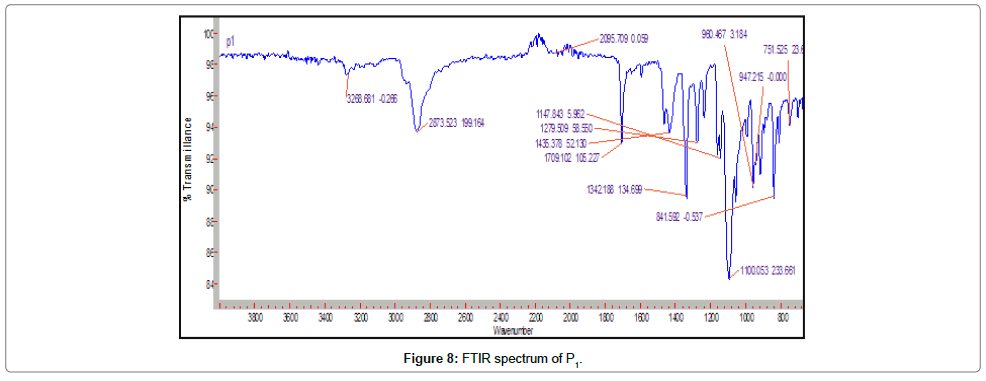
Figure 8: FTIR spectrum of P1.
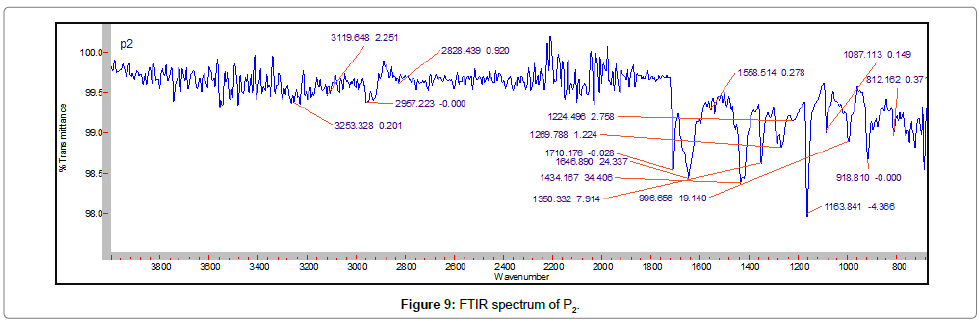
Figure 9: FTIR spectrum of P2.
Figure 10: FTIR spectrum of PE1.
Figure 11: FTIR spectrum of PE2
Figure 12: FTIR spectrum of S1.
Figure 13: FTIR spectrum of S2.
| Functional group | Reported waves number (cm-1) | Observed waves number (cm-1) |
|---|---|---|
| C=O (carbonyl group) | 1680-1760 | 1706.583 |
| C-H Stretch | 2850-3000 | 2935.339 |
| OH | 3200-3355 | 3272.435 |
| C-N Stretch | 1375-1450 | 1430.871 |
| -C=C- Aromatic ring | 1500-1600 | 1596.01 |
| Para substituted on benzene ring | 800-925 | 811 |
Table 8: Characteristics peaks observed in gliclazide.
Differential Scanning Calorimetry (DSC): Differential scanning calorimetry (DSC) is faze and reliable method to screen drug excipients compatibility and provides maximum information about the possible interactions.
DSC thermograms Figures 14 and 15 indicated a reduction in melting point changes of batch P1 as compared to the pure gliclazide probably due to size reduction. The melting point of unprocessed gliclazide was 198.48°C, whereas the melting point of batch P1 was 73.67°C (evaporation of absorbed water) and 191.37°C (drug melting point). In the P1 batch the endotherm of drug shifted towards lower temperature and peak was gradually reduced area. The lower melting point of the drug in the P1 was because of the melting point depression. As the intensity of endotherm was markedly decreased in P1, the faster dissolution rate of the drug from solid dispersion is attributing in the reduction in the crystallinity of the drug. Same as DSC thermograms Figures 16 and 17 indicated a reduction in melting point changes of batch S1 as compared to the pure gliclazide probably due to size reduction. The melting point of batch PE1 and S1 was found to be 171.19°C and 179.43°C. Crystallization inhibition is attributed to the entrapment of drug molecules in polymer matrix during solvent evaporation. The results also indicated that the drug and polymers are compatible.
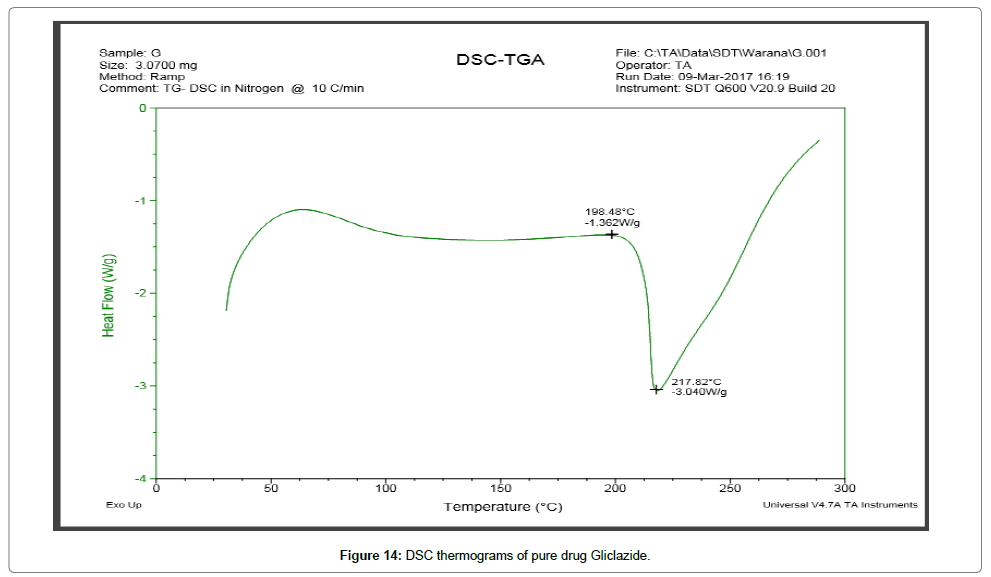
Figure 14: DSC thermograms of pure drug Gliclazide.
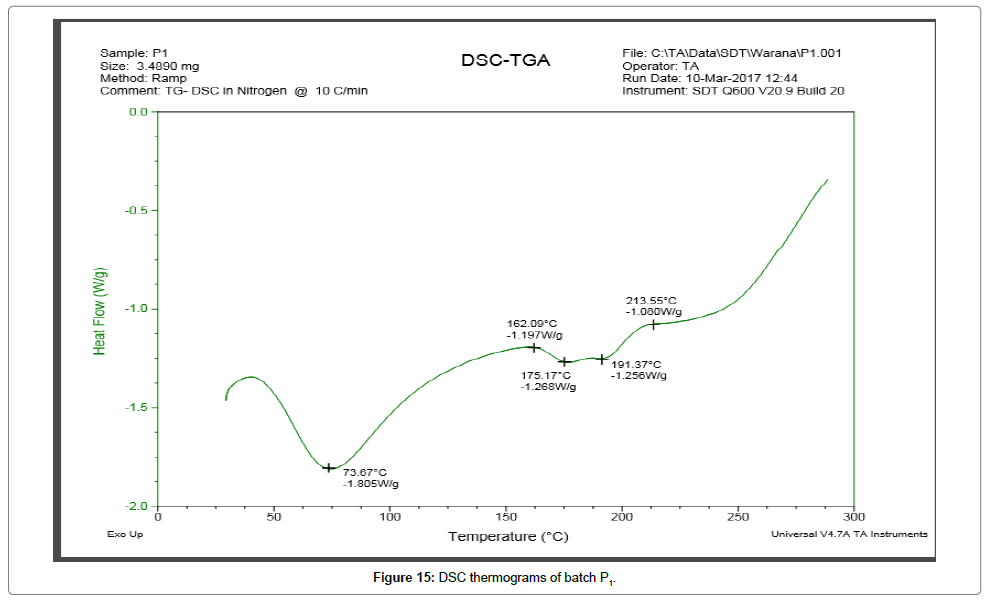
Figure 15: DSC thermograms of batch P1.
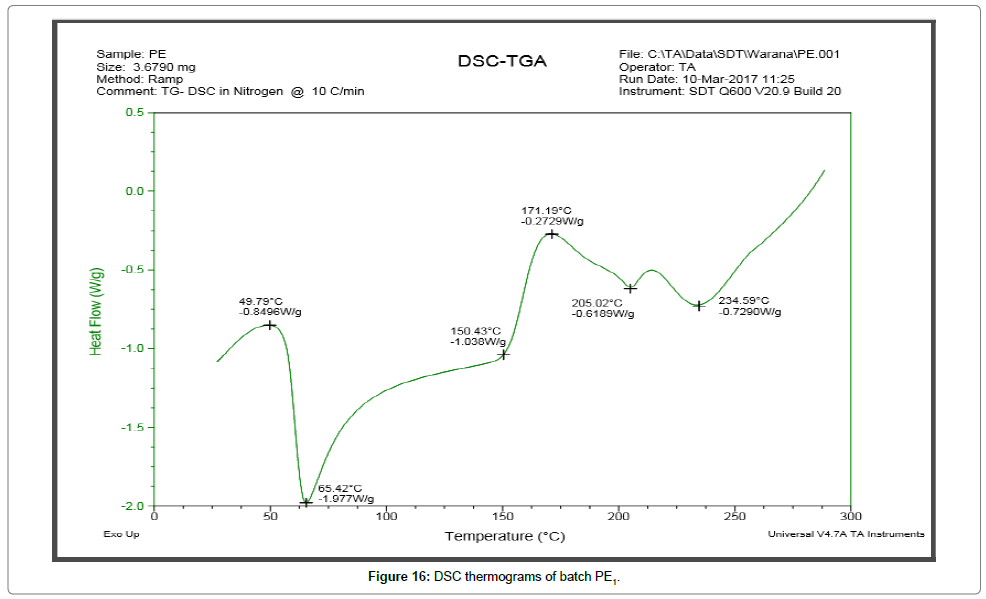
Figure 16: DSC thermograms of batch PE1.
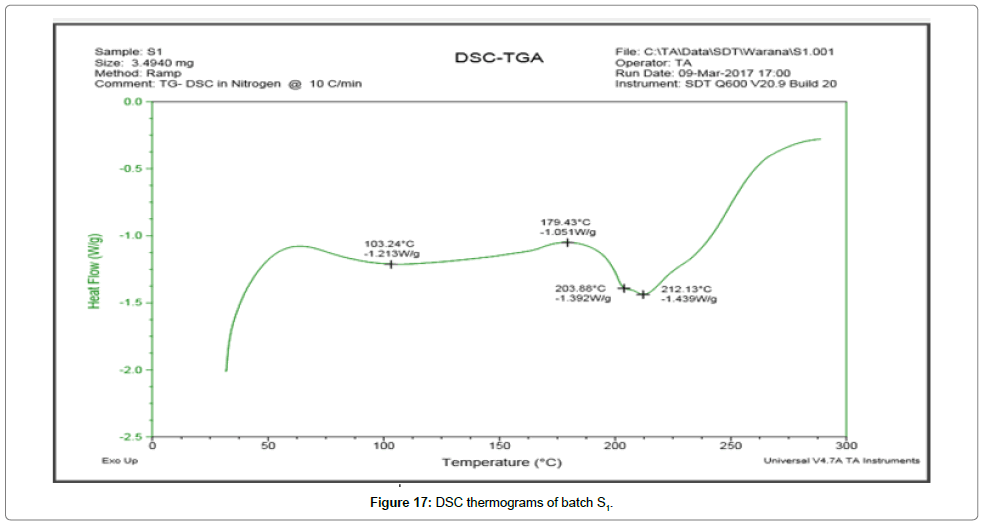
Figure 17: DSC thermograms of batch S1.
X-Ray Diffraction (XRD): The results of XRD was shown in Figure 18. In the XRD patterns highly intense and less diffused peaks shows the crystalline nature in the drug. The XRD patterns of pure gliclazide and batch P1 were very similar and showed sharp peaks at diffraction angles at 2θ of 10.77, 15.50, 17.90, 21.63, 23.42, 23.74 and 28.47°C. The peak heights for batch P1 and S1 were smaller, where PE1 having lager than those for the gliclazide. The peak height is affected by crystal size and crystallinity. The smaller peak heights of batch P1 was indicate that reduced crystal size and crystallinity as compared to gliclazide.
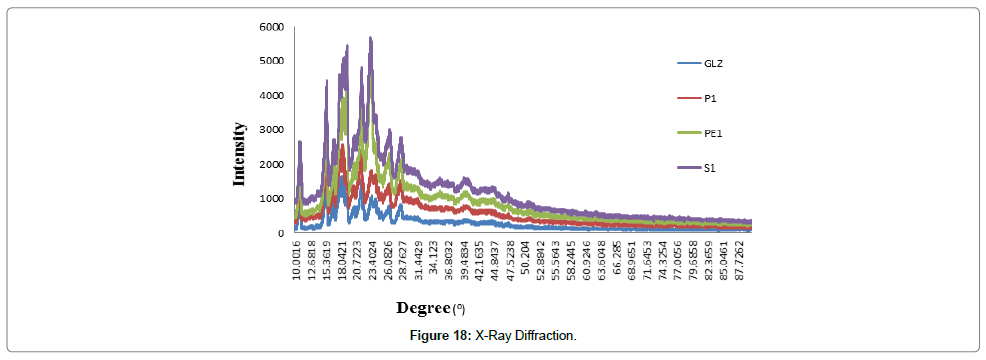
Figure 18: X-Ray Diffraction.
Scanning Electron Microscopy (SEM): Scanning Electron Microscope (SEM) images of pure gliclazide and batch P1 were obtained and are presented in Figure 19. Both pure gliclazide and batch P1 were crystalline and they showed different crystal habits. Crystals of unprocessed gliclazide were platy while crystals and batch P1 were prismatic and the prisms were elongated and looked like needles. These needle shape crystals could be attributed to presence of gliclazide in the crystalline state. The presence of carriers could inhibit crystals growth of GLZ during crystallization (solvent evaporation method). The effect of PVP as crystal growth inhibitor has been proved for GLZ are another drug before.

Figure 19: SEM images of a) pure gliclazide and b) batch P1.
Evaluation of Formulations
Pre-compression parameter of powder
The pre-compression parameter calculated as the formula given previously. The result was shown in Table 9. The Fast dissolving tablets of Gliclazide were prepared by using superdisintegrants- cross carmellose sodium and sodium starch glycolate at different concentrations alone and in combination by direct compression method. The powder blend was evaluated for Preformulation parameters and results are shown in Table 9. The angle of repose was in the range of 26.29°C-28.02°C indicating the good flow properties. Bulk density was found in the range of 0.55- 0.60 g/cm3 and the tapped density between 0.67-0.73 g/cm3. Hausner’s ratio was in the range of 1.169-1.218 indicating good flowability. The Carr’s compressibility index was found to be between 14.492-17.910%. Hence the prepared blends possessed good flow properties. The flow properties of powder were improved due to the decrease in the particle size which resulted in an increase in the specific surface area. The use of talc into the formulation will also help to improve flow properties.
| Formulation batches | Bulk density (ρb) gm/ ml | Tapped density (ρb) gm/ml |
Carr’s index % | Hausner’s ratio | Angle of repose (θ) |
|---|---|---|---|---|---|
| F1 | 0.56 | 0.67 | 16.41 | 1.19 | 28.02 |
| F2 | 0.55 | 0.67 | 17.91 | 1.21 | 26.55 |
| F3 | 0.60 | 0.73 | 17.80 | 1.21 | 26.81 |
| F4 | 0.60 | 0.71 | 15.49 | 1.18 | 27.71 |
| F5 | 0.58 | 0.68 | 15.04 | 1,18 | 26.29 |
| F6 | 0.57 | 0.68 | 16.17 | 1.19 | 27.10 |
| F7 | 0.59 | 0.70 | 15.64 | 1.18 | 26.96 |
| F8 | 0.59 | 0.69 | 14.49 | 1.16 | 27.84 |
Table 9: Precompression parameter of different tablet formulation.
Evaluation of fast dissolving tablet of gliclazide
The evaluation of fast dissolving tablet by various parameters was shown in Tables 10 and 11.
| Formulation batches | Hardness (N) (kg/cm2) Mean ± SD | Friability % Mean ± SD | Thickness(mm) Mean ± SD | Weight variation (mg) Mean ± SD | Disintegration time (sec) Mean ± SD |
|---|---|---|---|---|---|
| F1 | 3.53 ± 0.057 | 0.40 ± 0.01 | 3.06 ± 0.11 | 249 ± 1.15 | 20 ± 0.57 |
| F2 | 3.46 ± 0.115 | 0.40 ± 0.005 | 2.83 ± 0.57 | 250 ± 1.00 | 21 ± 1.15 |
| F3 | 3.50 ± 0.100 | 0.80 ± 0.228 | 2.76 ± 0.10 | 249 ± 1.00 | 16 ± 1.52 |
| F4 | 3.60 ± 0.200 | 0.53 ± 0.233 | 2.93 ± 0.15 | 250 ± 1.52 | 11 ± 1.00 |
| F5 | 3.60 ± 0.200 | 0.67 ± 0.225 | 3.28 ± 0.15 | 249 ±1.73 | 25 ± 1.15 |
| F6 | 3.56 ± 0.057 | 0.39 ± 0.005 | 3.03 ± 0.45 | 251 ± 1.00 | 18 ± 1.52 |
| F7 | 3.43 ± 0.057 | 0.53 ± 0.231 | 2.73 ± 0.20 | 249 ± 1.15 | 25 ± 0.57 |
| F8 | 3.30 ± 0.264 | 0.80 ± 0.005 | 2.83 ± 0.15 | 250 ± 1.52 | 22 ± 1.00 |
Table 10: Evaluation of fast dissolving tablet of gliclazide.
| Formulation batches | Wetting Time (Sec) Mean ± SD | Water absorption ratio (%) Mean ± SD |
Drug content (%) Mean ± SD |
Drug release (%) Mean ± SD |
|---|---|---|---|---|
| F1 | 23 ± 1.00 | 56.64 | 95.53 ± 0.894 | 46 ± 1 |
| F2 | 21 ± 1.15 | 58.46 | 94.24 ± 0.438 | 40 ± 3 |
| F3 | 19 ± 1.52 | 57.77 | 98.65 ± 0.502 | 32 ± 2 |
| F4 | 14 ± 1.00 | 58.22 | 99.67 ± 0.386 | 44 ± 4 |
| F5 | 24 ± 1.15 | 57.61 | 97.84 ± 0.444 | 38 ± 3 |
| F6 | 17 ± 1.00 | 57.04 | 99.16 ± 0.772 | 31 ± 2 |
| F7 | 23 ± 0.57 | 58.14 | 99.38 ± 0.889 | 38 ± 1 |
| F8 | 22 ± 0.57 | 58.09 | 98.10 ± 0.444 | 29 ± 2 |
Table 11: Evaluation of Fast Dissolving Tablet of gliclazide.
Wetting time
The wetting time for all the formulated tablets was in the range of 14 ± 1.000 to 24 ± 1.154 sec (Figure 20).

Figure 20: Tablet a) before wetted and b) after wetted.
Water absorption ratio
The water absorption ratio shows that in water get absorbed and tablet get dispersed. Water absorption ratio ranged from 056.64 ± 0.433 to 58.46 ± 0.230% (Figure 21).

Figure 21: Tablet a) before wetted and b) after wetted.
In vitro drug release studies for tablet
The percentage cumulative drug release of prepared formulation batches are shown in Figures 22 and 23 as for comparative study. The powder blend containing drug was compressed by using direct compression technique and Glicazide FDT’s were prepared. The compressed tablets were evaluated for physical properties and the results are tabulated in Tables 10 and 11, and the hardness was in the range of 3.30 ± 0.264 to 3.3.60 ± 0.200 kg/cm2. The tablet hardness was not more than 4.00 ± 0.200 kg/cm2 for best result. The thickness varies between 2.73 ± 0.18 to 3.06 ± 0.115 mm the smaller thickness of tablet shows good result. Uniformity of weight of prepared tablets was found within specifications of Indian Pharmacopoeia. Uniformity of weight was found to be in the range of 249 ± 1.000 to 251 ± 1.000 mg. The friability of all formulations was found to be less than 1.0% and was in the range of 0.39 ± 0.005 to 0.80 ± 0.005% indicating a good mechanical resistance of tablets. The disintegration time of all the formulated tablets was found to be in the range of 11 ± 1.000 to 25 ± 1.154 sec.
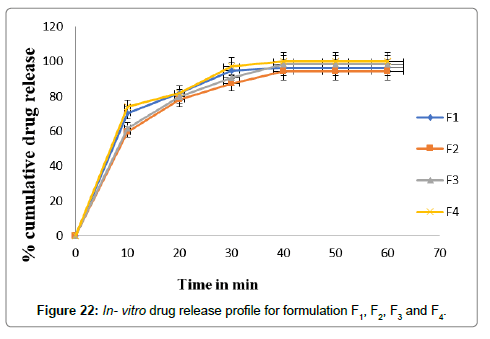
Figure 22: In- vitro drug release profile for formulation F1, F2, F3 and F4.
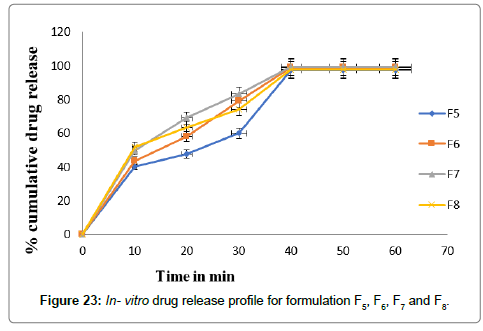
Figure 23: In- vitro drug release profile for formulation F5, F6, F7 and F8.
The formulation F4 shows lowest disintegration time i.e., 11 sec. The Percentage drug content was found in between 94.24 ± 0.438 to 99.38 ± 0.889%. All the formulations in vitro drug release results were mentioned in the Table 11. In vitro drug release of the prepared fast dissolving tablets was performed in pH 7.4 using USP dissolution apparatus type 2. The result of in vitro drug release was shown in Table 11.
From the result, it was concluded that fast dissolving tablets of Gliclazide using SDs dispersion batch P1 can be successfully prepared by direct compression techniques using different concentrations of super disintegrants cross carmellose sodium and sodium starch glycolate in combinations solvent evaporation method has successfully improve solubility and dissolution rate of gliclazide.