Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2017) Volume 10, Issue 11
The information on the 3D structure of a protein including its folding mechanism is encoded in its amino acid sequence. A β-trefoil protein is well known to have a remarkable 3D structure property, that is, the pseudo three-fold symmetry without clear hydrophobic packing. It is interesting to investigate how information on the folding mechanism to form such a topology is encoded in the amino acid sequence of a protein. In this study, analyses based on inter-residue average distance statistics and the conservation of hydrophobic residues are performed for sequences of 26 β-trefoil proteins to identify significant sites for the initial folding. Results are compared with the native 3D structures. The conserved hydrophobic residues are defined by multiple sequence alignment based on the 3D structures. It is confirmed that a conserved hydrophobic residue is always located in a β-strand. In particular, β-strands 5 and 6 are significant for the initial folding from the analyses based on the inter-residue average distance statistics. These results coincide well with the experimental data obtained so far for folding of some of the β-trefoil proteins. It is also confirmed that the conserved hydrophobic residues defined in this study contribute to form hydrophobic packing in β-trefoil proteins in general. Twelve conserved hydrophobic residue pairs are almost always observed to form packing in the 26 β-trefoil proteins from different superfamilies. We elucidate how the conserved hydrophobic residues in β-strands 5 and 6 contribute to the initial stage of folding of a β-trefoil protein. The common packing of the 12 conserved hydrophobic residue pairs are significant to form the whole β-trefoil fold structure.
Keywords: β-trefoil fold; Folding; Inter-residue average distance statistics; Multiple sequence alignment; Conserved hydrophobic residues
The 3D structure of a β-trefoil fold protein shows the remarkable property of a pseudo three-fold symmetry without clear hydrophobic packing, and it exists ubiquitously in the protein structure space as a member of the superfold proposed by Orengo et al. [1] exhibiting various functions. The first discovered example of such a protein with this fold was soybean trypsin inhibitor, the 3D structure of which was determined by Sweet et al. [2]. The specific properties of the 3D structure of this protein were described in detail by McLachlan [3]. Murzin et al. [4] proposed calling this specific 3D structure the “β-trefoil fold”.
Figure 1(a) represents an example of the 3D structure of a β-trefoil protein, that is, 29-kDa galactose-binding lectin with the PDB code of 2RST, and each structural unit or subdomain exhibiting the threefold symmetry is also shown in Figure 1(b). A schematic drawing of the topology of a β-trefoil protein consisting of 12 β-strands with intervening loops forming the trefoil shape is presented in Figure 1(c). The six β-strands, 1, 4, 5, 8, 9 and 12, form the barrel structure (β-strands colored orange) and the rest of the β-strands constitute three β-hairpins (hairpin triplets), 2-3, 6-7 and 10-11 (β-strands colored green).

Figure 1: (a) 3D structure of a protein with the β-trefoil fold (29-kDa galactose-binding lectin (PDB ID:2RST) as an example). β-Strands colored orange form a barrel structure and β-strands indicated by green color are so-called cap strands. (b) Fundamental structural unit in the β-trefoil fold. The red, green, and blue units are the segments 1-53, 54-89, and 90-132 in 2RST. (c) Schematic drawing of β-trefoil topology.
According to the SCOPe database, the β-trefoil fold can be grouped into eight superfamilies [5-7]. The sequence identity among proteins from different superfamilies in the β-trefoil fold is rather low as shown in Table 1. It is quite interesting how proteins from different superfamilies in the β-trefoil assemble into the same β-trefoil topology in spite of the rather low sequence identity. This suggests that only a small number of amino acids are determinant of the β-trefoil fold. Such amino acids may be conserved during evolution.
| Superfamily (PDB ID) | 2K8R | 2RST | 3BX1_D | 1HCD | 1T9F |
|---|---|---|---|---|---|
| Sequence identity (%) | |||||
| Cytokine (2K8R) | 9.0 | 7.4 | 12.1 | 6.3 | |
| Ricin B-like lectins (2RST) | 6.9 | 9.6 | 3.9 | ||
| STI-like (3BX1_D) | 4.9 | 10.1 | |||
| Actin-crosslinking proteins (1HCD) | 8.2 | ||||
| MIR domain (1T9F) | |||||
Table 1: Sequence identity among the selected proteins from five superfamilies, cytokine (PDB ID:2K8R), ricin B-like lectins (PDB ID:2RST), STI-like (PDB ID:3BX1_D), actin-crosslinking proteins (PDB ID:1HCD), and MIR domain (PDB ID:1T9F), in the β-trefoil fold.
Thus, a β-trefoil protein attracts the interests of many researchers. McLachlan [3] Murzin et al. [4] Ponting et al. [8] Lee et al. [9,10] Broom et al. [11,12] Longo et al. [13,14] and Xia et al. [15] pointed out that the β-trefoil structure might be constructed by triplication of the gene produced by gene duplication from a trefoil unit during evolution [3,4,8-15]. From the analyses of sequences and 3D structures of β-trefoil proteins by Murzin et al. [4] and Ponting et al. [8], and from the success to design protein sequences exhibiting the β-trefoil structure by Lee et al. [9,10] Broom et al. [11,12] Longo et al. [13,14] and Xia et al. [15], it is believed that the present β-trefoil proteins evolved from a common ancestor of a homotrimer protein. Longo et al. [13,14] and Xia et al. [15] revealed based on Φ-value analyses that folding nuclei of fibroblast growth factor 1 (FGF-1), that is, a β-trefoil protein, can be significant units in the initial stage of the evolution of β-trefoil proteins. They designed and produced several kinds of artificial β-trefoil proteins: a protein with three-repeat partial sequences, “Symfoil” using the Top- Down Symmetric Deconstruction method, a β-trefoil protein formed by a homotrimer called “Monofoil” [9,10,15], and a protein having the same folding nuclei to those in fibroblast growth factor 1 (FGF-1) detected by Φ-value analysis. The latter protein is called “Phifoil” [13-15].
Folding experiments of the following β-Trefoil proteins were performed extensively: fibroblast growth factor 1 (FGF-1), interleukin-1 beta (IL-1β) in cytokine superfamily and hisactophilin-1 (His) in the actin-crosslinking proteins superfamily. Φ-value analyses [13-15] for FGF-1, H/D exchange experiment by NMR [16-19] and Gō model simulations [20,21] for FGF-1, IL-1β and for His were performed to identify their folding mechanisms. These studies revealed the differences in overall folding pathways among these proteins, while these proteins commonly start to fold at the central β-strands [13,17].
Li et al. [22,23] and Feng et al. [24] conducted multiple sequence alignments of selected β-trefoil proteins based on their 3D structures and proposed key residues in the three symmetrical units to form the β-Trefoil fold.
How the information on the folding mechanism to form such the β-trefoil topology is encoded in the amino acid sequence of a protein is a very interesting problem. In the present study, we attempt to identify the significant residues to form the β-trefoil unit from their sequences by using a contact map and contact frequency prediction of a residue in a protein in random state based on inter-residue average distance statistics. These methods can predict the folding properties of proteins. We have confirmed so far that our techniques can extract information of folding mechanisms from the sequence of a protein for the following proteins: fatty acid binding proteins [25], globin-like fold proteins [26], IgG binding and albumin binding domains [27,28], immunoglobulin-like fold proteins [29], ferredoxin-like fold proteins [30], and lysozyme (to be published). Using these techniques significant parts for folding in an amino acid sequence can be detected with relatively high accuracy. Furthermore, we try to identify conserved hydrophobic residues in representative proteins in the β-trefoil fold. However, the sequence identities of these proteins are rather low, and making accurate multiple alignments based on only sequences is difficult. Thus we use information of the 3D structures to make multiple alignments. The information of conserved hydrophobic residues in combination with the results based on the average distance statistics is used to detect the significant residues to form the β-trefoil structure in spite of the low sequence identity. We also focus on hydrophobic packing formed by conserved hydrophobic residues in the native structures and compare the packing hydrophobic residues and the residues significant to folding.
Target proteins
We collected proteins with the β-trefoil fold according to the classification of SCOPe 2.05 [5-7]. Sequences in a same superfamily were aligned with MAFFT [31] and classified into several groups of sequences with more than 40% sequence identity. A sequence in a group was selected as the representative. That is a protein with one domain including no irregular structure up to around 180 residues and of which folding experiments and/or Gō model simulations were performed was selected as possible as we could. As a result, the 26 proteins belonging to 5 superfamilies (Table 2) were selected for the present study.
| Superfamily | PDB ID | UniProt ID | Protein name (UniProtKB) | Sequence length |
|---|---|---|---|---|
| Cytokine | 2K8R | P05230 | Fibroblast growth factor 1 | 133 |
| 1Q1U | P61328 | Fibroblast growth factor 12 | 138 | |
| 2FDB_M | P55075 | Fibroblast growth factor 8 | 147 | |
| 1QQK | Q02195 | Fibroblast growth factor 7 | 129 | |
| 1J0S | Q14116 | Interleukin-18 | 157 | |
| 6I1B | P01584 | Interleukin-1 beta (Homo sapiens [Human]) | 153 | |
| 1MD6 | Q9QYY1 | Interleukin-36 receptor antagonist protein | 154 | |
| 2KKI | P01583 | Interleukin-1 alpha | 151 | |
| 2WRY | O73909 | Interleukin-1 beta (Gallus gallus [Chicken]) | 155 | |
| 2P39 | Q9GZV9 | Fibroblast growth factor 23 | 142 | |
| 2P23 | O95750 | Fibroblast growth factor 19 | 136 | |
| Ricin B-like lectins | 2RST | O96048 | 29-kDa galactose-binding lectin | 132 |
| 1SR4_A | O06522 | Cytolethal distending toxin subunit A | 167 | |
| 1SR4_C | O06524 | Cytolethal distending toxin subunit C | 154 | |
| 1KNM | P26514 | Endo-1,4-beta-xylanase A | 129 | |
| 1DQG | Q61830 | Macrophage mannose receptor 1 | 134 | |
| STI-like | 3BX1_D | P07596 | Alpha-amylase/subtilisin inhibitor | 181 |
| 1TIE | P09943 | Trypsin inhibitor DE-3 | 166 | |
| 1R8N | P83667 | Kunitz-type serine protease inhibitor DrTI | 185 | |
| 1WBA | P15465 | Albumin-1 | 171 | |
| 2GZB | P83051 | Kunitz-type proteinase inhibitor BbCI | 164 | |
| 3ZC8 | D2YW43 | Trypsin inhibitor | 182 | |
| 3TC2 | Q8S380 | KTI-B protein | 181 | |
| Actin-crosslinking proteins | 1HCD | P13231 | Hisactophilin-1 | 118 |
| MIR domain | 1T9F | O61793 | Protein R12E2.13 (Caenorhabditis elegans) | 176 |
| 3HSM | P11716 | Ryanodine receptor 1 | 164 |
Table 2: Target proteins in this study.
Analyses with inter-residue average distance statistics. Contact map analysis based on inter-residue average distance statistics
We present a brief summary to construct a contact map from the inter-residue average distance statistics. This map is called an average distance map (ADM). The details are described in refs [32,33] and supplementary material. The inter-residue average distances and standard deviations were calculated using known 3D structures taking the amino acid types and sequence separation into account.
The separation of two residues along the sequence was taken into consideration as follows: M=1 when 1 ≤ k ≤ 8, M=2 when 9 ≤ k ≤ 20, M=3 when 21 ≤ k ≤ 30, M=4 when 31 ≤ k ≤ 40, and so on, where k=|i-j| and M is called as range. The average distances of all pairs of amino acid types were calculated in each range. A plot is made on a map, when the average distance of a pair of amino acids in a range M is less than a threshold value set beforehand. An ADM for a protein can be constructed from only its sequence in this way. A region with high density of plots can be regarded as a region predicted to be compact in a 3D structure. “η-value” of a region is used as a measure of compactness. We regard a predicted compact region by ADM with high an η-value as a compact region in the early stage of folding.
So far, it has been confirmed that the predicted compact regions by ADMs for myoglobin and plant leghemoglobin [25], fatty acid binding proteins [25] and ferredoxin-like fold [30] proteins correspond well to the early folding regions detected by NMR studies [34,35], kinetic studies [36] and Φ-value analyses [37-40].
Contact frequency analysis using a potential derived from the present inter-residue average distance statistics
Using a potential derived from the present inter-residue average distance statistics, contact frequency of a residue was calculated in a random state to identify a residue where initial folding events, such as hydrophobic collapse, occur [25]. We employed a Cα bead model to represent a structure of a protein in this study. For a simulation of protein conformations, the Metropolis Monte Carlo method with the potential energy εi,j derived from average distance ͞ri,j and its standard deviation σi,j was employed. The bond and dihedral angles of the initial conformation were randomly selected. The bond and dihedral angles between the residue i and i+1 is bent and rotated randomly and followed during the simulation by Metropolis judgment to determine whether the new conformation is accepted or not. That is, we perform a simulation from a totally random distribution with the restriction derived from the average distance statistics.
One step includes alteration of all the bond and dihedral angles followed by the Metropolis judgment. We assume that the probability density with the potential energy between two residues, 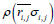 , is equal to the probability density derived from the standard Gaussian distribution calculated with its average distance and standard deviation, , as follows:
, is equal to the probability density derived from the standard Gaussian distribution calculated with its average distance and standard deviation, , as follows:
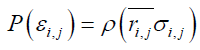 (1)
(1)
where this equation is expressed by equation (2);
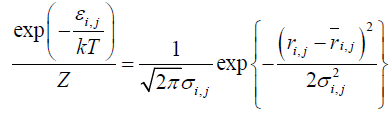 (2)
(2)
Equation (2) leads the equations (3) and (4):
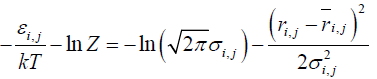 (3)
(3)
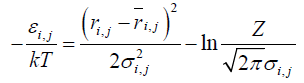 (4)
(4)
where kT is set so that the acceptance ratio is 0.5. A significant value in a calculation is the difference between the energy values of conformations, and Z does not appear in the calculation explicitly. Thus, Z is ignored in the calculations.
It is expected that we can obtain ensembles with reproducible inter- residue average distance statistics using this potential. The contact frequency, g(i, j), for each pair of residues is estimated with structures generated from a simulation using the potential energy function. Then the residue contact frequency, g(i, j), in the same range M is normalized as follows:
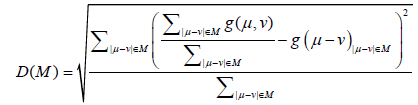 (5)
(5)
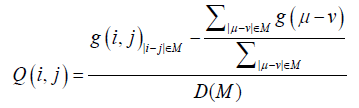 (6)
(6)
where μ or v is the residue number.
Finally, the relative contact frequency, Fi, is obtained by summing the normalized contact frequencies, Q(i, j), from j=1 to N for each residue i, where N is the total number of residues:
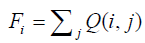 (7)
(7)
The value Fi is called the F-value. Residues at peaks in the plot of F-values are expected to be located in the center of many inter-residue contacts, such as a hydrophobic cluster. That is, a region near a peak of an F-value plot is likely to be important for folding, especially for its initial stage. Ten simulations with 60000 steps were performed, calculating the average of the F-values for residue i. (30 simulations may be required for a credible statistic. In our case, because of the computational cost, we made just 10 simulations for each case.) We calculate the sampled structure from the very beginning of the simulation. A peak is defined when the difference in the values of a valley and a peak is more than the following cut-off value, Fcut;
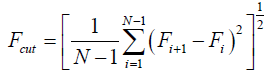 (8)
(8)
where Fi is the F-value of residue i and N is the total residue number.
It has been confirmed that a hydrophobic residue near the F-value plot for a protein tends to form hydrophobic packing in the native structure of a protein for IgG binding domain [27] and ferredoxin-like fold proteins [30].
Structure-based multiple sequence alignment
A multiple sequence alignment taking 3D structures into account by means of Combinatorial Extension [41] program in STRAP software [42] was used to detect the relationships among sequences with relatively low sequence identity.
Conserved hydrophobic residues and hydrophobic packing
Considering the significance of interactions between hydrophobic residues, conserved hydrophobic residues were identified based on the result of a structure-based multiple sequence alignment for sequences with low sequence identity. The following residues Ala, Val, Leu, Ile, Met, Phe, Trp and Tyr are regarded as hydrophobic. (Tyr is included in the hydrophobic residues because Phe and Tyr are mutatable to each other as observed in the PAM matrix [43]). When more than 80% of residues at an aligned site are those residues, hydrophobic residues at this site are regarded as being conserved. The definition of hydrophobic packing is based on the reduction of solvent accessible surface area (SASA). That is, when the packing of two hydrophobic residues make a reduction of SASA by more than 10Å2, these hydrophobic residues are regarded as forming hydrophobic packing. The calculation of SASA was performed with the Shrake-Rupley Algorthm [44].
Structure-based sequence alignment and conserved hydrophobic residues
Figure 2(a) indicates the result of the structure-based multiple sequence alignment of 26 sequences shown in Table 1 by means of STRAP, in order to identify the conserved hydrophobic residues and elucidate the common regions by ADM predictions. The sequence identities of these proteins are rather low (Table 1), so making the accurate multiple alignment is difficult. Thus we use information of the 3D structures to make multiple alignment. The result of ADM prediction for each protein is shown in Figure 2(a) (Figures S1-4 in supplementary material). A predicted compact region is indicated by a red bar. Brighter red denotes higher η-value. The conserved hydrophobic residues in the alignment are indicated by a yellow letter in a predicted compact region and by a blue letter out of a predicted compact region in Figure 2(a). (By the examination of the correctness of the alignment of the β-strands of these proteins, we could confirm the correct alignment of whole sequences.) Table 1 presents the sequence identities showing around 25- 35% identity within the same superfamily but only about 10% identity between proteins from different superfamilies. However, almost all pairs of β-trefoil proteins exhibit RMSD of about 3Å indicating high similarity of their 3D structures in spite of the low sequence identities (Table S1 in supplementary material).

Figure 2: (a) Structure-based sequence alignment for 26 β-trefoil proteins with the results of ADM predictions. A predicted compact region is indicated by a red bar. Brighter red denotes higher η-value. The color code is presented in the top of the figure. The predicted region with the highest η value of a protein is shown by the brightest red color and the region with lowest brightness means the lowest ηvalue in this protein. The conserved hydrophobic residues in the alignment are indicated by a yellow letter in a predicted compact region and by a blue letter out of a predicted compact region (b, c) The histogram of the ratio of the residues included in predicted compact regions by ADMs to the number of residues aligned at a site. A region with high histogram values denotes the high conservation of compact region. A red line denotes 70% conservation. An arrow indicates the location of a β-strand in 2RST as an example. (The fine figure is also presented in Figure S4 in supplementary material).
Fifteen conserved hydrophobic residues are identified, and they appear in every β-strand as shown in Figure 2(a). We label these 15 conserved hydrophobic residues by the numbers of the β-strands. For example, the conserved hydrophobic residue in the β-strand 1 is labeled by CHR-β1. The β-strands 2, 4, and 5 contain two conserved hydrophobic residues, respectively. In the case of the β-strand 2, a conserved hydrophobic residue located in the N-terminal side is labeled as CHR-β2N and in the same way that in the C-terminal side is labeled as CHR-β2C. We confirm that these conserved hydrophobic residues correspond well to the conserved residues proposed by Murzin et al. [4] and Feng et al. [24]. Murzin et al. defined the conservation of hydrophobic residues in two proteins from the Cytokine superfamily and one protein from STI-like superfamily. Thus, multiple alignment of sequences from various superfamilies leads to the similar results. Feng et al. already performed the structure-based sequence alignment for β-trefoil fold proteins. We think that a similar result is obtained in our present study, and our results reveal clearer conservation by focusing on hydrophobic residues Ala, Val, Leu, Ile, Met, Phe, Trp and Tyr.
Figure 2(b) shows a histogram denoting the conserved compact regions predicted by ADMs (details to make it are shown Figure S3 in supplementary material). Although we can always make a rough correspondence of the location of the predicted compact regions by ADMs to the actual positions of the trefoil units for each protein, there is variety in the predicted patterns of the predicted compact regions. Figure 2(b) seems to exhibit this situation. However, the histogram indicates some conserved regions of the compact regions. That is, β-strands 1-3, β-strand 4, β-strand 5, β-strand 6, and β-strands 8-11 are in the regions with more than 70% conservation as shown in Figure 2(b). The threshold of 70% conservation is indicated by a red line at the histogram in Figure 2(b). These regions are corresponding moderately well to Trefoil-1, Trefoil-2 and Trefoil-3, respectively.
Although a portion with low conservation between β-strands 7 and 8 in Trefoil-2 is observed in Figure 2(b), this portion is a relatively long loop and seems not to be conserved during evolution.
ADM and F-value analyses for β-trefoil proteins of which folding mechanisms have been experimentally investigated
In this section, we treat fibroblast growth factor 1 (FGF-1) (PDB ID: 2K8R), interleukin-1 beta (IL-1β) (PDB ID:6I1B), and hisactophilin-1 (His) (PDB ID:1HCD), because these proteins have been investigated extensively to reveal their folding mechanisms by experimental techniques.
Fibroblast growth factor 1 (FGF-1) (2K8R): FGF-1, also called heparin-binding growth factor 1, is classified in the fibroblast growth factors family in the cytokine superfamily in SCOPe. This protein binds to heparin at the residues 105-121, called turn 11 [13,14,18]. It has been reported that this functional site folds in the late stage of the folding process (the foldability-function tradeoff hypothesis) [13,14,21]. In preceding studies, residues in β-strand 2 and β-strands 5-8 are protected in the early stage of folding revealed by H/D exchange experiments of NMR [17,18]. Longo et al. [13-14] and Xia et al. [15] reported that the folding nucleus detected by the Φ-value analyses consists of residues 16- 58 in β-strands 2-6. Figures 3 and 4 presents the ADM and the F-value plot with the results of the H/D exchange experiment for 2K8R.

Figure 3: Average Distance Map (ADM) for fibroblast growth factor 1 (FGF- 1, PDB ID:2K8R). This map predicts four compact regions 6-49 (η=0.350), 57-67 (η=0.214), 76-91 (η=0.226), and 100-128 (η=0.204). A black bar and a black arrow along the diagonal denote an α-helix and a β-sheet, respectively. A green bar means the position of a trefoil unit.

Figure 4: (a) F-value plot for fibroblast growth factor 1 (FGF-1, PDB ID:2K8R) with the result of ADM (the black bars near the abscissa). The residues on the peaks of this profile are denoted by open circles. A β-strand is indicated by a bold arrow with a numeral. The highest 5 residues are marked by black arrows. These residues are located around the β-strands 5-7. The histogram shown in this figure means the protection factor values obtained by the H/D exchange experiment [18]. (b) F-value plot for fibroblast growth factor 1 (FGF-1, PDB ID:2K8R) with the result of ADM (the black bars near the abscissa) with the positions of conserved hydrophobic residues indicated by black triangles. In both figures, we plot a value of F+ σ for each residue as a red line and F-σ as a blue line, where σ means the standard deviation.
We use PDB ID to distinguish proteins in this paper. The compact regions predicted by the ADM are residues 6-49 corresponding to β-strands 1-5, residues 57-67 corresponding to β-strands 6-7, residues 76-91 corresponding to β-strands 8-9, and residues 100-128 corresponding to β-strands 10-12. It is noticed that the predicted compact regions 6-49, 57-67 and 76-91, and 100-128 roughly correspond to the first, second, and third trefoil units, respectively. We refer the predicted compact regions 6-49, 57-67, 76-91 and 100-128 as region-1, region-2, region-3 and region-4. In particular, the predicted compact region 6-49 (a region colored red in Figure 5(a)) shows the highest η of 0.350, and this is expected to form stable compact structure in the initial stage of folding.

Figure 5: 3D structure of fibroblast growth factor 1 (FGF-1, PDB ID:2K8R). (a) Predicted compact region 6-49 by ADM colored red. (b) Folding nucleus obtained by the φ-value analysis [13-15] indicated by the purple region. (c) The residues with high protection factor values in the H/D exchange experiment [18] are colored red, purple, or orange in the order from high to low protection factor values in the space-filling model.
As recognized in Table 3, for 2K8R, all conserved hydrophobic residues are contained in the predicted compact regions (the number of all conserved hydrophobic residues is 15; the number of the residues within the predicted compact regions is 100 and the total number of residues is 133 as indicated in Table 3).
| PDB ID | Sequence length | Total number of residues in the predicted compact regions by ADM | Ratio of total number of residues in the predicted compact regions by ADM to the total number of residues | Number of conserved hydrophobic residues in the predicted compact regions by ADM | Ratio of the number of conserved hydrophobic residues in the predicted compact regions by ADM to the total number of conserved hydrophobic residues | Number of conserved hydrophobic residues out of the predicted compact regions by ADM | Total number of packing formed by conserved hydrophobic residues |
|---|---|---|---|---|---|---|---|
| 2K8R | 133 | 100 | 0.75 | 15 | 1.00 | 0 | 35 |
| 1Q1U | 138 | 106 | 0.77 | 14 | 0.93 | 1 | 43 |
| 2FDB_M | 147 | 82 | 0.56 | 12 | 0.80 | 3 | 40 |
| 1QQK | 129 | 71 | 0.55 | 10 | 0.67 | 5 | 40 |
| 1J0S | 157 | 124 | 0.79 | 15 | 1.00 | 0 | 28 |
| 6I1B | 153 | 124 | 0.81 | 14 | 0.93 | 1 | 38 |
| 1MD6 | 154 | 112 | 0.73 | 13 | 0.87 | 2 | 37 |
| 2KKI | 151 | 111 | 0.74 | 13 | 0.87 | 2 | 38 |
| 2WRY | 155 | 104 | 0.67 | 11 | 0.73 | 4 | 40 |
| 2P39 | 142 | 114 | 0.80 | 13 | 0.87 | 2 | 39 |
| 2P23 | 136 | 93 | 0.68 | 13 | 0.87 | 2 | 40 |
| 2RST | 132 | 83 | 0.63 | 11 | 0.73 | 4 | 39 |
| 1SR4_A | 167 | 102 | 0.61 | 10 | 0.67 | 5 | 36 |
| 1SR4_C | 154 | 125 | 0.81 | 14 | 1.00 | 0 | 30 |
| 1KNM | 129 | 74 | 0.57 | 10 | 0.67 | 5 | 37 |
| 1DQG | 134 | 114 | 0.85 | 14 | 0.93 | 1 | 38 |
| 3BX1_D | 181 | 124 | 0.69 | 14 | 0.93 | 1 | 36 |
| 1TIE | 166 | 86 | 0.52 | 12 | 0.80 | 3 | 36 |
| 1R8N | 185 | 130 | 0.70 | 13 | 0.87 | 2 | 38 |
| 1WBA | 171 | 139 | 0.81 | 15 | 1.00 | 0 | 37 |
| 2GZB | 164 | 89 | 0.54 | 12 | 0.80 | 3 | 37 |
| 3ZC8 | 182 | 117 | 0.64 | 11 | 0.73 | 4 | 39 |
| 3TC2 | 181 | 125 | 0.69 | 14 | 0.93 | 1 | 36 |
| 1HCD | 118 | 74 | 0.63 | 13 | 0.87 | 2 | 29 |
| 1T9F | 176 | 106 | 0.60 | 10 | 0.67 | 5 | 39 |
| 3HSM | 164 | 107 | 0.65 | 15 | 1.00 | 0 | 37 |
Table 3: Statistics of conserved hydrophobic residues.
It is interesting that this region corresponds well to residues 16-58 that were identified as the folding nucleus by Long et al. [13,14] and Xia et al. [15] as shown in Figure 5(b) (in this figure the folding nucleus obtained by the Φ-value analysis [13-15] is indicated by the purple region and in Figure 5(c) the residues with high protection factor values in the H/D exchange experiment [18] are colored red, purple, and orange in the order from high to low protection factor values in the space-filling model.) Thus, it is expected that a region predicted by ADM with the highest η value forms a stable compact or a structured region in the early stage of folding.
The high peaks in the F-value plot for this protein appear around β-strands 5-7 as presented in Figure 4(a). This area coincides with the highly protected region measured by NMR, that is, β-strands 5-8 as shown in Figure 4(a) (histogram indicated by black bars). A peak in the F-value plot appeared in this region is close to a peak of the histogram of the H/D protection factors within three residues as presented in Table 4. Therefore, the peaks in the F-value plot can be considered as a site to be structured in the early stage of folding based on the comparison with the H/D exchange experiment in this protein. It should be noted that the conserved hydrophobic residues CHR-β5N, CHR-β5C, CHR-β6 and CHR-β7 are near the peaks of the F-value plot as shown in Table 4 and Figure 4(b). Considering this result, the predicted compact region 57-67 by ADM can be regarded as a compact region in the early stage of folding. It is also considered that this part will be stabilized by the interactions with probably 6-49.
| Highly protected residues in the H/D exchange experiment | Residues at the highest peaks in the F-value plot | Difference in the sequence | Conserved hydrophobic residues near a peak in the F-value plot | Difference in the sequence |
|---|---|---|---|---|
| SER51 | TYR48 | 3 residues | VAL47 (CHR-β5N) ILE49 (CHR-β5C) |
4 residues 2 residues |
| TYR57 | GLN56 | 1 residue | LEU58 (CHR-β6) | 1 residue |
| ALA59 | 2 residues | |||
| GLY64 | ASP63 | 1 residue | LEU66 (CHR-β7) | 2 residues |
| GLY68 | LEU66 | 2 residues | LEU66 (CHR-β7) | 2 residues |
Table 4: Highly protected residues in the H/D exchange experiment [18] and the residues at the highest peaks in the F-value plot for 2K8R.
Next, packing formed by conserved hydrophobic residues for each protein is examined. In Figure 6, the following description applies. A conserved hydrophobic residue is indicated by a number or a number with N or C. Conserved hydrophobic residues near the highest peak in the F-value plot are placed in the blue cells. A black circle means a contact, and a red circle means a contact formed by a residue in a blue cell with a residue in a different predicted compact region. Conserved hydrophobic residues near the highest peak in the F-value plot tend to form contacts with conserved hydrophobic residues within various predicted compact regions and also linking predicted compact regions.

Figure 6: A kind of contact map showing the contacts between conserved hydrophobic residues in 2K8R. “Region-1” means the first predicted compact region by ADM for 2K8R.
For 2K8R, as shown in Figure 4(b), the highest peaks in the F-value plot are around CHR-β5N, CHR-β5C, CHR-β6 and CHR-β7. CHR- β5N and CHR-β5C are in the predicted compact region 6-49 by ADM, and CHR-β6 and CHR-β7 exist in the predicted compact region 57-67. Figure 6 shows a kind of contact map for just the conserved hydrophobic residues. That is, a plot is made when two conserved hydrophobic residues form a packing. The residues near the peak in the F-value plot, CHR-β5N, CHR-β5C, CHR-β6 and CHR-β7 form contacts widely from region-1 to region-4 (these contacts are indicated by a red circle).
Wang and Yu [18] demonstrated in their study that β-strands 5, 6 and 7 (including Val47, Tyr48, lle49, Tyr57, Leu58, Leu65, Leu66 and Tyr67) form a hydrophobic core with Tyr57, Leu58 and Ala59 as center of the hydrophobic core in the native FGF-1 structure. Our prediction shows that the center of folding of 2K8R is CHR-β5N, CHR-β5C, CHR-β6 and CHR-β7 with the conserved hydrophobic residues Val47 (CHR-β5N), lle49 (CHR-β5C), Leu58 (CHR-β6) and Leu66 (CHR-β7) and coincides well to the results of Wang and Yu. It is noted that the heparin binding site of this protein, 105-121, includes only a few conserved hydrophobic residues near a peak in the F-value plot, and this fact seems reflect that this part is not strongly involved in the folding indicating the foldability-function tradeoff hypothesis [13,14,21].
Interleukin-1 beta (IL-1β) (6I1B): This protein is a kind of inflammatory cytokine and classified to the interleukin-1 family of the cytokine superfamily according to SCOPe. The studies of the H/D exchange experiments [16,19] were done previously. According to these studies, protection of β-strands 6-10 occurs in the early stage of folding followed by closure of the barrel structure formed by β-strands 1-4, 11- 12 [16,19].
In the present study, the region 3-73 (region-1) includes β-strands 1-6 with η-value of 0.262 and the region 99-151 (region-2) including β-strands 8-12 with that of 0.321 suggesting that the region 99-151 is more stable in the early stage folding as shown in Figures 7 and 8. That is, the first predicted compact region corresponds to Trefoil-1 and the N-terminus of Trefoil-2, whereas the second predicted compact region corresponds to the C-terminus of Trefoil-2 and Trefoil-3.

Figure 7: Average Distance Map (ADM) for interleukin-1 beta (IL-1β, PDB ID:6I1B). This map predicts two compact regions 3-73 (η=0.262), and 99-151 (η=0.321). A black bar and a black arrow along the diagonal denote an α-helix and a β-sheet, respectively. A green bar means the position of a trefoil unit.

Figure 8: (a) F-value plot for interleukin-1 beta (IL-1β, PDB ID:6I1B) with the result of ADM (the black bars near the abscissa). The residues on the peaks of this profile are denoted by open circles. A β-strand is presented by a bold arrow with a numeral. The highest 5 residues are marked by black arrows. These residues are located around β-strands 4 and 5-6. (b) F-value plot for interleukin-1 beta (IL-1β, PDB ID:6I1B) with the result of ADM (the black bars near the abscissa) with the positions of conserved hydrophobic residues indicated by black triangles. In both figures, we plot a value of F+σ for each residue as a red line and F-σ as a blue line, where σ means the standard deviation.
The predicted compact region by ADM for 6I1B contains 14 conserved hydrophobic residues out of 15 (the number of the residues within the predicted compact regions is 124, and the total number of residues is 153) as indicated in Table 3.
The highest peak is on β-strand 6 as shown in Figure 8(a). According to Liu et al. [16] and Capraro et al. [19], the folding of 6I1B occurs at β-strands 6-10, and our results show that β-strands 8-12 would be stable in the early stage of folding (the η-value of the region 99-151 including β-strand 8-12 is higher) reflecting the results of Liu et al. and Capraro et al., although β-strand 6 is included in the first compact region. The highest peak in the F-value plot is in β -strand 6 indicating the strong involvement of this β-strand in folding. Thus, we consider that the significant parts for the folding in this protein are region 99-151 and β-strand 6, and this speculation corresponds to the result of Liu et al. and Capraro et al. The peaks in the F-value plot are all included in the predicted compact regions by ADM.
The conserved hydrophobic residues CHR-4N, CHR- β4C, CHR- β5N, CHR- β5C and CHR6 are near the peaks of the F-value plot for 6I1B as shown in Table S2 and Figure 8(b).
The packing formed by these conserved hydrophobic residues is presented in Figure 9. This figure shows that CHR-β4N, CHR-β4C, CHR-β5N, CHR-β5C and CHR- β6 form packing within the region 3-73 (region-1) and also with the conserved hydrophobic residues in region-2, CHR- β8, CHR- β9 and CHR- β10, indicating the significance of these residues for the 3D structure formation of 6I1B (these contacts are indicated by a red circle). It should be noted that these residues are near the moderately high peaks in the F-value plot. As mentioned earlier, CHR-β6 in region-1 may interact with the conserved hydrophobic residues in region-2, and it is confirmed that CHR-β6 forms hydrophobic packing with CHR- β8 and CHR- β10 in region-2.

Figure 9: A kind of contact map showing the contacts between conserved hydrophobic residues in 6I1B. “Region-1” means the first predicted compact region by ADM for 6I1B. Conserved hydrophobic residues near the highest peak in the F-value plot are located in region-1. These residues form packing within region 3-73 (region-1) and also with the conserved hydrophobic residues in region-2 indicating the significance of these residues for the 3D structure formation.
Hisactophilin-1 (His) (1HCD): Hisactophilin-1 (His) is a kind of actin binding protein, and its sequence contains 31 histidines out of total 118 residues, that is, this is a histidine-rich protein in comparison with other β-trefoil proteins. Another characteristic of this protein is that it contains shorter loops and β-strands compared with other β-trefoil proteins [16]. This is classified into the histidine-rich actin-binding protein (hisactophilin) family in the actin-crosslinking proteins superfamily in the SCOP database.
H/D exchange experiments and Gō model simulations were also performed for this protein so far by Liu et al. [16] and Chavez et al. [20]. The results of the H/D exchange experiments show that this protein folds at β-strand 4-8 at the beginning and β-strands 1-3 and 10-12 are structured in the end of the folding [16], while Gō model simulations suggest that the folding proceeds at the central β-strands (Trefoil-2) and the C-terminal region (Trefoil-3) [20].
Figure 10 presents that the predicted compact regions by ADM involve residues 5-36 (η=0.146, region-1) including β-strands 1-4, 43- 63 (η=0.210, region-2) including β-strands 5-7, and 73-93 (η=0.135, region-3) and including β-strands 8-10. These three predicted compact regions roughly correspond to Trefoil-1-3. The central predicted compact region 43-63 exhibits the highest η-value and is expected to be stable in the early stage of folding.

Figure 10: Average Distance Map (ADM) for hisactophilin-1 (His, PDB ID:1HCD). This map predicts three compact regions 5-36 (η=0.146), 43-63 (η=0.210), and 73-93 (η=0.135). A black bar and a black arrow along the diagonal denote an α-helix and a β-sheet, respectively. A green bar means the position of a trefoil unit.
The predicted compact region by ADM for 1HCD includes 13 (the number of the residues within the predicted compact regions is 74 and the total number of residues is 118).
The F-value plot of hisactophilin shows the highest peak at the β-strand 6 in the second predicted compact region as shown in Figure 11, suggesting the frequent contact formations in the early stage of folding. The results of both the ADM and the F-value plot analyses reflect those of the H/D exchange experiments that indicate the formation of the β-strand 4-8 at the beginning.

Figure 11: (a) F-value plot for hisactophilin-1 (His, PDB ID:1HCD) with the result of ADM (the black bars near the abscissa). The residues on the peaks of this profile are denoted by open circles. A β-strand is presented by a bold arrow with a numeral. The highest 5 residues are marked by black arrows. These residues are located around the β-strands 5-7 and 9. (b) F-value plot for hisactophilin-1 (His, PDB ID:1HCD) with the result of ADM (the black bars near the abscissa) with the positions of conserved hydrophobic residues indicated by black triangles. In both figures, we plot a value of F+σ for each residue as a red line and F-σ as a blue line, where σ means the standard deviation.
The conserved hydrophobic residues CHR-β5N, CHR-β5C, CHR-β6 and CHR-β9 are near the peaks of the F-value plot as shown in Table S3 and Figure 11(a).
Our analyses and experimental studies reveal that the folding mechanism of FGF-1 is different from those of IL-1β, that is, the folding proceeds from the central to N-terminal regions in FGF-1, whereas IL- 1β folds from the central to C-terminal regions [16]. In any case, these three proteins fold from the central β-strands. As we see, the present analyses of ADMs and F-value plots show good correspondence with the results of the experimental study. Furthermore, the conserved hydrophobic residues near the peaks of the F-value plots are considered to be significant for the folding. Therefore, it is confirmed that we can regard a region predicted by ADM as a significant part for folding in the β-trefoil proteins in this study. Thus, it is demonstrated that the folding properties of these three β-trefoil proteins can be predicted from their amino acid sequences. In the present work, we further perform the ADM for other 23 β-trefoil proteins and the F-value analyses for 3 selected β-trefoil proteins from respective superfamilies in addition to the information on conservation of hydrophobic residues to infer their folding mechanisms of these proteins.
As shown in Figure 11(b), the conserved hydrophobic residues near the highest peaks are CHR-β5N, CHR-β5C, CHR-β6, CHR-β7 and CHR-β9. CHR-β5N, CHR-β5C and CHR-β6 in the predicted compact region 43-63 (region-2) with the highest η-value and CHR-β9 is in region 73-93 (region-3). The packing formed by the conserved hydrophobic residues in the native structure is presented Figure 12.

Figure 12: A kind of contact map showing the contacts between conserved hydrophobic residues in 1HCD. “Region-1” means the first predicted compact region by ADM for 1HCD.
CHR-β5C and CHR-β6 form hydrophobic packing within the region 43-63 and also the conserved hydrophobic residues interact each other connecting the predicted compact regions as observed in Figure 12. Again, the significance of the conserved hydrophobic residues for folding is suggested.
ADM and F-value analyses for 29-kDa galactose-binding lectin, alpha-amylase/subtilisin inhibitor and protein R12E2.13 from C. elegans
The three proteins examined so far are from two superfamilies. In this section, we select three proteins from the rest of the superfamilies, 29-kDa galactose-binding lectin (2RST), alpha-amylase/subtilisin inhibitor (3BX1) and protein R12E2.13 from C. elegans (1T9F). We try to make predictions based on the present techniques for these proteins. Experimental data on folding have not yet been reported for them.
2RST (29-kDa galactose-binding lectin): The results of ADM analysis with the F-value plot for 29-kDa galactose-binding lectin (2RST) are shown in Figure 13. ADM predicts regions 30-63 including β-strands 3-6 (η=0.190, region-1) and 83-131 including β-strands 8-12 (η=0.379, region-2). Figure 13 suggests that the C-terminal part would be stable in early stage folding because of the larger η-value. The predicted compact regions for 2RST include 11 conserved hydrophobic residues as shown in Table 3 (the number of the residues within the predicted compact regions is 83 and the total number of residues is 132). The high peaks in the F-value plot appear around CHR-β5N, CHR-β5C, CHR-β6, CHR-β8, CHR-β9 and CHR-β10. The peaks around CHR- β5N, CHR-β5C, and CHR-β6 are again observed in this protein as the same for the other three proteins (fibroblast growth factor 1 (FGF-1), interleukin-1 beta (IL-1β) and hisactophilin-1 (His)) suggesting these residues are significant for the initial folding. (We take a conserved hydrophobic residue within ± 5 residues from a peak. The reason of “within ± 5 residues” is as follows: a peak in the F-value plot for 2K8R is close to a peak of the histogram of the H/D protection factors within ± 3 residues, and a conserved hydrophobic residue always exists near a peak of the H/D protection factor histogram for 2K8R within ± 4 residues in Table 4. From these facts, we take “within ± 5 residues” as a threshold.) The regions around CHR-β5N, CHR-β5C and CHR-β6 are included in the conserved predicted compact regions as shown in Figure 2(b).

Figure 13: F-value plot for 29-kDa galactose-binding lectin (PDB ID:2RST) with the result of ADM (the black bars near the abscissa) with the positions of conserved hydrophobic residues indicated by black triangles. A β-strand is shown as a bold arrow with a numeral. The high peaks in the F-value plot appear around CHR-β5N, CHR- β5C, CHR- β6, CHR- β8, CHR- β9 and CHR- β10 indicated by arrows. We plot a value of F+σ for each residue as a red line and F-σ as a blue line, where σ means the standard deviation.
Among the conserved hydrophobic residues in 2RST, CHR-β5N, CHR-β5C, CHR-β6 CHR-β8, CHR-β9 and CHR-β10 are near the highest peaks as presented in Figure 13. CHR-β5N, CHR-β5C and CHR-β6 are in the predicted compact region 30-63 (region-1) and CHR-β8, CHR-β9, CHR-β10 are including the predicted compact region 83-131 (region-2). Those conserved hydrophobic residues make the hydrophobic interactions within region-1 and region-2 or between these two regions as shown in Figure 14. Thus, the conserved hydrophobic residues in these regions are considered to be significant for the 3D structure of this protein.

Figure 14: A kind of contact map showing the contacts between conserved hydrophobic residues in 2RST. “Region-1” means the first predicted compact region by ADM for 2RST.
3BX1 (alpha-amylase/subtilisin inhibitor): Figure 15 presents results of ADM analysis with the F-value plot for alpha-amylase/ subtilisin inhibitor (3BX1). The predicted compact regions by ADM are regions 6-47 including β-strands 1-3 (η=0.217, region-1) and 60- 68 including β-strand 4 (η=0.157, region-2) and 73-110 including β-strand 5-7 (η=0.181, region-3) and 141-175 including β-strand 9-12 (η=0.266, region-4). The predicted compact regions for 3BX1 include 14 conserved hydrophobic residues as shown in Table 3 (the number of the residues within the predicted compact regions is 124 and the total number of residues is 181 in Table 3). It is suggested from this figure that the C-terminal 141-175 would be stable in early stage folding because of the larger η-value. The highest peak in the F-value plot appears around CHR-β5N, CHR-β5C and CHR-β6 as for the previous three proteins. Region 73-110 would also form a stable compact region but would be weaker compared to the region 141-175. The residues around the peaks on CHR-β5N, CHR-β5C and CHR-β6 may interact with the residues in the region 141-175 and region 73-110 may merge with region 141-175. These regions are included in the conserved predicted compact regions as shown in Figure 2(b).

Figure 15: F-value plot for alpha-amylase/subtilisin inhibitor (PDB ID:3BX1_D) with the result of ADM (the black bars near the abscissa) with the positions of conserved hydrophobic residues indicated by black triangles. A β-strand is presented by a bold arrow with a numeral. The high peaks in the F-value plot appear around CHR-β5N, CHR-β5C and CHR-β6 indicated by arrows. We plot a value of F+σ for each residue as a red line and F-σ as a blue line, where σ means the standard deviation.
Among the conserved hydrophobic residues in 3BX1, the highest peaks of the F-value plot are observed around CHR-β5N, CHR-β5C and CHR-β6 (Figure 15). These three residues are in the predicted compact region 73-110 (region-3). Figure 16 indicates that these three conserved hydrophobic residues form packing with other conserved hydrophobic residues in every predicted compact region.

Figure 16: A kind of contact map showing the contacts between conserved hydrophobic residues in 3BX1_D. “Region-1” means the first predicted compact region by ADM for 3BX1_D.
1T9F (protein R12E2.13 from C. elegans): Figure 17 indicates the results of ADM analysis with the F-value plot for protein R12E2.13 from C. elegans (1T9F). Regions 4-41 cover β-strands 1-3 (η=0.303, region-1) and 47-69 cover β-strand 4 (η=0.192, region-2) and 115-159 cover β-strand 8-11 (η=0.191, region-3). The predicted compact regions for 1T9F include 10 conserved hydrophobic residues as shown in Table 3 (the number of the residues within the predicted compact regions is 106 and the total number of residues is 176). The ratio of the conserved hydrophobic residues in the predicted compact regions to those in the whole sequence is 0.67, whereas the ratio of the number of residues within the predicted compact regions to the number of residues in the whole sequence is 0.6. Thus, the conserved hydrophobic residues tend to be included in the predicted compact regions also in this protein. It is suggested from Figure 17 that the N-terminal 4-41 would be stable in the early stage folding because of the larger η-value, and region 47-69 may merge with the N-terminal region to fold. The high peaks appear around CHR-β4N, CHR-β4C, CHR-β5C, CHR-β6 and CHR-β8. One high peak appears also around CHR-β5C and CHR-β6 as in the previous three proteins. These regions around these peaks are included in the conserved predicted compact regions as shown in Figure 2(b).

Figure 17: F-value plot for protein R12E2.13 from Caenorhabditis elegans (PDB ID:1T9F) resulting from ADM (the black bars near the abscissa) with the positions of conserved hydrophobic residues indicated by black triangles. A β-strand is shown by a bold arrow with a numeral. The high peaks in the F-value plot appear around CHR-β4N, CHR-β4C, CHR-β5N, CHR-β5C, CHR-β6 and CHR-β8 indicating by arrows. We plot a value of F+σ for each residue as a red line and F-σ as a blue line, where σ means the standard deviation.
Among the conserved hydrophobic residues in 1T9F, the conserved hydrophobic residues near the highest peaks of the F-value plot are CHR-β4N, CHR-β4C, CHR-β5C, CHR-β6 and CHR-β8 (Figure 17). CHR-β4N and CHR-β4C are included in the predicted compact region 47-69 (region-2) and CHR-β8 is in the predicted compact region 115- 159 (region-3), but any predicted compact region does not contain CHR-β5C and CHR-β6. Figure 18 presents the packing formed by these conserved hydrophobic residues. These five conserved hydrophobic residues form packing with other conserved hydrophobic residues.

Figure 18: A kind of contact map showing the contacts between conserved hydrophobic residues in 1T9F. “Region-1” means the first predicted compact region by ADM for 1T9F.
These results suggest the significance of a conserved hydrophobic residue near a peak in an F-value plot to make packing to connect predicted compact regions to form a whole protein 3D structure. The packing formed by conserved hydrophobic residues in 2K8R, 6I1B, 1HCD, 2RST, 3BX1 and 1T9F is presented in Figure S5 and 6.
In the present study, we performed the structure-based sequence alignment for β-trefoil proteins and conserved hydrophobic residues were identified. Interestingly, every β-strand contains one or two conserved hydrophobic residues in spite of low sequence identity among sequences. This fact may indicate that these equally distributed hydrophobic residues need to form the symmetrical β-trefoil fold. Similar results were already obtained by Murzin et al. [4] and Feng et al. [24], and they revealed the relationships between conserved residues and 3D structures or interaction energies. Feng et al. [24] also found “symmetric key structural residues” specific for the β-trefoil structure based on structure-based multiple sequence alignment of domains in five two-domain proteins with two β-trefoil structures. Furthermore, they elucidated that these symmetric key structural residues are well-conserved in the majority of β-trefoil proteins. Conserved hydrophobic residues identified in our study correspond well to the symmetric key structural residues obtained by Feng et al. indicating our conserved hydrophobic residues are also key residues to make packing within the β-trefoil fold as discussed later.
Furthermore, we demonstrated that the results of ADM and F-value analyses reflect the results of folding experiments for proteins 2K8R, 6I1B and 1HCD. Longo et al. [13,14] succeeded in designing Phifoil based on a folding nucleus of 2K8R deduced from the results of the η-value analyses, and this folding nucleus comprises β-strand 2 to β-strand 6. This nucleus corresponds to the ADM predicted compact regions 6-49 and 57-67 which includes some of highest peaks in the F-value plot. That is, our predicted compact regions correspond well to the folding nucleus defined from φ-value analyses. For 6I1B, the present study predicts the C-terminal part is significant for the folding reflecting the experimental data [16,19], although the highest peak in the F-value plot is included in the N-terminal predicted compact region by ADM. The region around this peak is also included in the folding region identified by Liu et al. [16] and Capraro et al. [19].
The present study performed the analyses by the ADMs in the combination with the structure-based sequence alignment, and the results predict the compact regions stable in the early stage of folding for β-trefoil proteins. Although compact regions of β-trefoil proteins look to show a variety of locations of the predicted regions, the modest conservation of the compact regions is also observed as shown in Figure 2(b). The fact that the predicted stable compact regions with the highest η value for 2K8R, 6I1B and 1HCD correspond well to the experimentally obtained folding units denotes a predicted compact region by ADM can be regarded as a kind of unit of folding in a β-trefoil protein. Based on this, it is considered that the results in Figure 2(b) indicate the variety of the folding mechanisms of the β-trefoil proteins. On the other hand, from the F-value analyses β-strands 5 and 6 are always the center of folding for 2K8R, 6I1B and 1HCD consistent with the experimental results. This property is always observed in the results of the F-value plots for other β-trefoil proteins for which folding mechanisms have not yet been examined experimentally. Further folding mechanism, that is, with which part, N-terminal or C-terminal part, β-strands 5 and 6 interact, depends on each protein.
For 2K8R the high peaks in the F-value plot correspond well to the high peaks in the histogram of the protection factors [18]. Considering the significance of conserved hydrophobic residues for the formation of the 3D structure of a β-trefoil protein, this result indicates that a conserved hydrophobic residue near a high peak in an F-value plot is important for hydrophobic packing in the early stage of folding. CHR-β5N, CHR-β5C and CHR-β6 always appear near the highest or second highest peak in an F-value plot in a protein from almost every superfamily (Table S3). That is, CHR-β5N, CHR-β5C and CHR-β6 are considered as commonly significant residues to start folding in β-trefoil proteins.
Thus, we consider that the present analyses can be applied to predict the folding properties for β-trefoil proteins in general. Furthermore, the information of the location of conserved hydrophobic residues in combination with the results of ADM and an F-value plot provides the significant residues for hydrophobic packing during folding.
The number of packing residue pairs for each protein is around 30~40. From Table 3, the conserved hydrophobic residues tend to be included in the predicted compact regions by ADMs. This indicates that the interactions between them tend to occur within a compact region or between compact regions.
Figure 19 shows the number of observed contacts between the conserved hydrophobic residues in β-strands for 26 proteins examined in this study. Contacts between CHR-β1 and CHR-β2N, CHR-β4C and CHR-β5C, CHR-β5C and CHR-β8, CHR-β6 and CHR-β8, CHR-β8 and CHR-β9, CHR-β8 and CHR-β10, and CHR-β9 and CHR-β10 appear in 26 proteins (Figure 19, highlighted by a color). Contacts between CHR-β1 and CHR-β4N, CHR-β4N and CHR-β5C, CHR-β5C and CHR-β6, CHR-β6 and CHR-β7 and CHR-β7 and CHR-β10 also appear in 25 proteins (Figure 19, highlighted by a color). Proteins not showing some of these contacts are 1J0S and the C chain of 1SR4. These contacts are those between CHR-β1 and CHR-β4N, CHR-β4N and CHR-β5C, CHR-β5C and CHR-β6, CHR-β6 and CHR-β7 and CHR-β7 and CHR-β10. We take them into consideration since misalignment is observed for 1J0S and the C chain of 1SR4. Thus, these 15 contacts seem to be especially significant for β-trefoil folding. They, including contacts formed by CHR-β5C and CHR-β6 are mainly formed within Trefoil-2 and some conserved hydrophobic residues in Trefoil-1 and Trefoil-3 as shown in Figures 19 and 20. That is, the significance of Trefoil-2 including CHR-β5C and CHR-β6 is suggested. It is worth noting that the common hydrophobic contact between CHR-β6 and CHR-β8 is especially significant to connect region-1 and region-2 in 6I1B as pointed out earlier.

Figure 19: The number of contacts formed by each conserved hydrophobic pair in 26 β-trefoil proteins. Trefoil-1, 2 and 3 are indicated by red, green and blue, respectively. A contact within each unit is indicated by a color specific to Trefoil-1, 2 or 3. A contact formed between units is indicated by yellow.

Figure 20: Common hydrophobic packing formed by conserved hydrophobic residues in the 26 β-trefoil proteins appeared in 2K8R expressed by the space-filling model. Three trefoil units are colored by red, green and blue.
Information on the location of conserved hydrophobic residues in combination with the results of ADM and an F-value plot reveal the significant residues for hydrophobic packing during folding. In other words, an initial folding site in a protein can be defined as a site with conserved hydrophobic residues near a high F-value peak in a predicted region by ADM with the highest η value. Several such sites form contacts within a predicted region by ADM and form a larger structure.
Although every β-strand contains one or two conserved hydrophobic residues and these equally distributed hydrophobic residues seem to be significant to form the symmetrical β-trefoil fold, the conserved hydrophobic residues in Trefoil-2 may be more significant. CHR-β5N, CHR-β5C and CHR-β6 contain conserved hydrophobic residues near the highest or second highest peak of an F-value plot in a protein from almost every superfamily. As mention earlier, the results of folding experiments for several β-trefoil proteins indicates the significance of β-strands 5 and 6 for folding. Combining our present’s results, CHR- β5C and CHR-β6 are considered to be the center of β-trefoil formation in general.
It is worth reiterating a point in our Introduction. Once again we see in the proteins studied in this paper that folding is a consequence of amino acid sequence [45].
This work was supported by JSPS KAKENHI Grant Number 26330335.