Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2014) Volume 2, Issue 6
Thrombocytopenia has remained a major concern in post-haematopoietic stem cell transplant patients. It is a strong negative indicator for survival and is often seen in patients with advanced acute and chronic Graft versus host disease (GVHD). The pathogenesis of thrombocytopenia varies from impaired thrombopoiesis to increased turnover of platelets. The dose of CD34+ cells and that of megakaryocyte specific lineage cells (CD34+ /CD41+ and CD34+ / CD61+ ) has a positive effect on thrombopoiesis. Conditions like bone marrow fibrosis, CMV infection and ganciclovir administration affect the bone marrow environment leading to ineffective thrombopoiesis. Increased destruction of platelets is seen with GVHD, drugs, infections, thrombotic microangiopathy and platelet refractoriness due to alloimmunization. The management of thrombocytopenia includes treating the underlying conditions and supportive care with platelet transfusion. Platelet refractoriness is managed using ABO matched platelets, HLA-matched platelets or cross-matched platelets for transfusion depending on the underlying cause. Some of the recent studies have concentrated on the effect on thromobopoiesis due to maturation of megakaryocytes or ploidy and the effect of cytokines on bone marrow microenvironment. Further studies are needed to incorporate these parameters as part of routine laboratory investigation in thromobocytopenic patients along with estimation of anti-platelet antibodies for better diagnosis and management of these patients.
Keywords: Thrombocytopenia; Hematopoietic Stem Cell; Ganciclovir
Conditioning chemotherapy and radiotherapy followed by hematopoietic stem cell transplantation (HSCT) is a successful treatment strategy for a variety of conditions like hematologic malignancies, aplastic anemia, congenital immunodeficiencies and inborn errors of metabolism [1]. The source of haematopoietic stem cells (HSC) can be autologous or allogeneic as per the indications for transplant. Stem cells are usually derived from marrow or mobilized from peripheral blood [2]. Profound thrombocytopenia is generally seen in all patients following myeloablative conditioning, which is due to the bone marrow aplasia caused by high-dose chemotherapy and/or total body irradiation (TBI). Following hematopoietic stem cell transplantation (HSCT), restoration of normal numbers of blood leukocytes and platelets usually follows predictable kinetics within the first month post-transplant, with increasing numbers of granulocytes, platelets, lymphocytes, and finally erythrocytes in the blood.
Prolonged thrombocytopenia is generally seen as a late complication in patients after allogenic stem cell transplation. It is mainly attributed to the source of stem cells, the doses of infused CD34cells, graft-versus-host disease (GVHD), engraftment failure, recurrence of the underlying malignancy, microangiopathy, alloimmunisation, drugs, or viral infection [3]. Two main theories are proposed for the pathogenesis of thrombocytopenia after SCT, first although there may be normal platelet production, peripheral destruction may occur due to anti-platelet autoantibodies, splenic sequestration, or other factors [4]. Secondly platelets production may be insufficient due to impaired thrombopoiesis [5]. However, in most clinical conditions there is an overlap of the two mechanisms [6]. With prophylactic platelet transfusions, serious bleeding is rare in the absence of significant post-transplant complications like infection, graft-versus-host disease (GVHD) or veno-occlusive disease (VOD) of the liver. Clinical manifestations vary as per the cause of the thrombocytopenia. This review will attempt to analyze the causes of thrombocytopenia after transplantation and also trying to study various characteristics associated with it.
The differential diagnosis and treatment of thrombocytopenia is problematic in many post-HSCT patients. The pathogenesis of thrombocytopenia has not fully been understood, although various mechanisms have been described.
Impaired platelet production
Impaired platelet production after HSCT has been associated with the dose of the CD34+ cells, relapse or persistence of disease; persistent marrow fibrosis; graft-versus-stroma effect, and drug-induced inhibition of marrow (Figure 1).
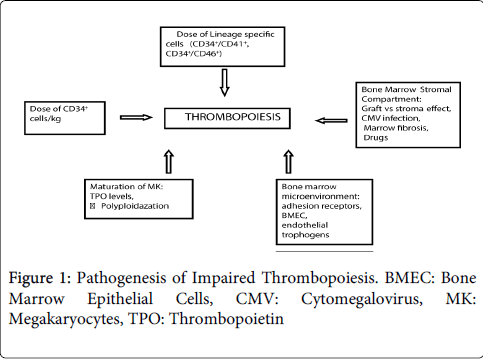
Figure 1: Pathogenesis of Impaired Thrombopoiesis. BMEC: Bone Marrow Epithelial Cells, CMV: Cytomegalovirus, MK: Megakaryocytes, TPO: Thrombopoietin
The progenitor cells or the totipotent stem cells develop into pluripotent or the multipotent stem cells. It is these cells which show the early expression of CD34+ markers. These pluripotent cells develop into the multipotent cells from which develop the various lineage cells viz myeloid, lymphoid, erythroid, megakaryocytic. All these cells express the CD34+ marker.
The lineage specific cells in addition express the co-markers specific to their cell lineages eg. myeloid (CD13 and 33), megakaryocytic (CD 41 and 61), erythroid (CD 71), B-lymphoid (CD19) and T-lymphoid (CD7) [7].
Although the cell marker specific to the pluripotent stem cell has not yet been identified, the progenitor cells which express the CD34+ cell marker are known to be capable of bone marrow engraftment. The relationship between CD34+ cell dose and engraftment is well documented [8]. There is a relationship between high CD34+ cell dose and better long-term haematopoiesis. A dose of >5 x 106 CD34+ cell/kg was shown to be the threshold dose between rapid and delayed haematopoietic engraftment in autologous transplant patients [7]. Even in the allogenic transplants, the CD34+ cell dose is shown to be the difference between early and delayed engraftment. Along with the dose of CD34+ cells the proportion of lineage specific cells in the total number of CD34=cells plays an important role [8]. Dercksen et al. showed that in patients who receied a higher dose of CD34+/CD41+ cells/kg the time for median platelate recovery was lower compared to those who received a lower dose. Another study by Woo et al. showed that dosage of CD34+/CD41+ cells, CD34+/CD61+ cells and that of Colony Forming Unit-MK (CFU-MK) positively affected the platelet recovery [9].
Bone marrow (BM) has two functional compartments viz haematopoietic and stromal. The haematopoietic compartment is radiation sensitive while the stromal compartment is radiation resistant. The stromal compartment further comprises of reticular stromal cells (RSC) and adipocytes. These provide a conducive environment and facilitate the engraftment and proliferation of the haematopoietic cells in the host [10]. Upon infusion when they enter the circulation, the haematopoietic stem cells of both bone marrow as well as peripheral blood origin, are recognised in the marrow and get anchored in the microenvironment of the bone marrow. They are also helped by their adhesive nature and their reactions with different stromal cells in this endeauvre. The sites at which the circulating HSC anchor themselves are provided by the endothelial galactosyl receptors and a heparan sulphate component of the extracellular matrix. There are adhesion receptors present on the CD34+ cells which play a pivotal role in engraftment of these cells in bone marrow microenvironment. A few of them which have been described are CXC chemokine receptor 4 (CXCR4), very late antigen (VLA)-4 (CD49d), VLA-5 (CD49e), lymphocyte function-associated antigen (LFA) -1, CD44. A correlation between expression of these adhesion molecules and time to platelet recovery has been documented in autologous transplant patients [11]. The engraftment of these cells in non-medullary sites is prohibited by inhibitory glycoproteins called restrictins.
The bone marrow microenvironment is an essential factor in this process. It consists of bone marrow epithelial cells (BMEC), endosteal cells and perivascular cells. The megakaryocytes (MK) also release endothlial trophogens like Stromal Derived Factor-1(SDF-1) and Vascular Endothelial Growth Factor (VEGF) which influence the viability of the BM microenvironment. VEGF is a proangiogenic factor which promotes angiogenesis through its receptors VEGF-R on BMEC. SDF-1 is responsible for promoting motility of MK and helps them to localise in the BMEC [12]. It thus promotes thrombopoiesis. Interference with this vascular microenvironment or interference with the motility of MK affects the thrombopoiesis. Reduction of BMEC, perivascular cells, SDF-1 and VEGF is seen in patients with prolonged thrombocytopenia compared to those wih good graft function and healthy donors. Also, the BMECs and MK-derived factors from the transplanted HSC play a role in influencing neovascularisation of the host marrow and in turn affect the viability of the engrafted MK [12].
After initial engraftment of the HSC a secondary thromobocytopenia is seen due to cytomegalovirus (CMV) infections. CMV infection interferes with reticular stromal cells. It interferes with haematopoiesis by creating a deficiency of the stroma-derived growth- and differentiation- promoting hemopoietins and interference at the stage of clonal [10]. Once engrafted in the marrow, the different lineage specific cells start regenerating. Platelets are derived from megakaryocytes. The mature MK contain a single, lobulated, polyploid nucleus and abundant cytoplasm. Platelets are derived from the cytoplasmic protrusions from the cell surface. This state of polyploidy is achieved through a process of DNA endomitosis. Upon maturation MK stop the process of proliferation, however, they keep increasing their DNA content without attaining the final stages of mitosis. This process is essential for the multilobation of nucleus and increase in the cytoplasmic volume. Although the polyploidazation does not interfere with the differentiation of MK, it is however essential for the production of platelets [13]. The immature MK show lower ploidy indicating a left shift. This study by Zhang et al. also established the presence of left shift in MK in patients of allo-HSCT compared to the healthy donors. In those post-HSCT patients with prolonged thrombocytopenia the left shift of MK was more pronounced. The total number of MK present in patients with and without thrombocytopenia did not differ much
It has been studied that levels of thrombopoietin (TPO), a cytokine responsible for megakaryogenesis and thrombpoiesis, are considerably increased in patients who showed prolonged thrombocytopenia compared to those who did not [13]. TPO considerably increases when the megakarocytes are very low or absent in the bone marrow. The levels of TPO in the blood are regulated by the level of its receptor c-Mpl which is expressed on the megakaryocytes [6]. The persistence or relapse of the primary disease is usually because the conditioning regimen including chemotherapy and TBI were unable to eradicate the disease [7]. Patients with severe marrow fibrosis before HSCT have a slower platelet recovery and higher requirements of platelet transfusion compared to those who do not have marrow fibrosis [14]. After allogeneic transplant, there is a graft-versus-stroma effect which may lead to poor marrow function resulting in slower platelet recovery. Drugs causing during peri transplant period may lead to delayed platelet engraftment or secondary failure, these include amongst others methotrexate, and trimethoprim – sulfamethoxazole [15].
Increased Platelet Destruction or Turnover
GVHD, VOD of liver and infections are the common complications after SCT leading to increased platelet turnover. The incidence of chronic GVHD is high in patients with long term survival post- HSCT. There is dysregulation of the host immune system which results in immunodeficiency and autoimmune changes. T-cells from the donor HSC play a major role in the immunological changes taking place in the host. Platelet autoantibodies have been detected in patients of chronic GVHD which cause premature removal of platelets from the circulation. Thrombocytopenia has been postulated as a marker of increased complications in patients of chronic severe GVHD [16]. In a study by R Yamazaki et al, in cases of prolonged thromobocytopenia, the glycocalicin index (GCI) was found to be very high compared to those with normal platelet counts. Glycocalicin is a cleaved fragment of a glycoprotein (GP) Iba which is specific to platelets. Also important is the detection of the auto-antibodies reactive with platelet surface GPs like GPIIb-IIIa which is a hallmark of immune thrombocytopenia, especially Idiopathic Thrombocytopenic Purpura. The B-cells which produce these antibodies are also increased [6].
VOD of the liver is a common complication of conditioning therapy. There is injury to hepatocytes and sinusoidal endothelial cells due to drugs like busulphan and cyclophosphamide [17,18]. Clinically it presents with tender hepatomegaly, ascites, coagulopathy and jaundice. Pathologically it involves the terminal hepatic venule leading to necrosis of hepatocytes, perivenular fibrosis and engorgement of sinusoids.VOD is associated with persistent thrombocytopenia and platelet transfusion refractoriness. Although the exact mechanism of the thrombocytopenia in VOD is unknown, there is platelet destruction in VOD.
Infections like herpes/adeno viruses can present with varied manifestations from skin vesicles, lung manifestations to neurologic complications. Parvoviruses commonly present with features of myelosuppression mainly anemia and flu like illness. All these infectious etiology with few others are very common in HSCT population and are frequently associated with thrombocytopenia. In most cases the effects that these have on platelets are successfully managed with appropriate preventive or early antibiotic or antiviral therapy.
Thrombotic microangiopathy (TMA) syndromes occur with a frequency of 0.5-76%. It generally presents as hemolytic uremic syndrome (HUS) or thrombotic thrombocytopenic purpura (TTP), which are well-recognized complications of SCT [19,20]. The classic presentation of TMA includes anemia, thrombocytopenia, hemolysis, raised LDH and acute renal dysfunction. Neurologic deficits may occur in up to half the cases. TMA usually occurs within the first 100 days after stem cell infusion.
George JN described TTP and HUS to be similar in clinical presentation except for the renal involvement in the latter [21]. The diagnosis of TTP is made in the presence of clinical features like thrombocytopenia and microangiopathic hemolytic anemia. Other clinical features like fever, renal and neurological abnormalities are shared with other complications post-HSCT. Many of the suspected TTP cases post-HSCT have systemic infections or acute GVHD. V Roy et al. described this condition as ‘TTP like syndrome [22]. They had higher risks for transplant-related complications, severe acute GVHD and other systemic infections. It causes diffuse endothelial damage by a plasma factor which results in apoptosis of endothelial cells. There is also a deficiency of a plasma enzyme which cleaves the von Willebrand factor (VWF).
Transfusion related challenges generally occur in patients requiring chronic platelet transfusions. Platelet refractoriness is defined as repeated failure to obtain satisfactory responses to platelet transfusions [23,24] and generally occurs in 13-100% of multi-transfused patients [25]. It is broadly classified as immune and non-immune [25,26] (Table 1). Immune causes are primarily because of Human Platelet Antigen (HPA) and Human Leukocyte Antigen (HLA) incompatibility. Non-immune causes are thought to be the major contributors to platelet refractoriness [27]. The major cause for immunologic refractoriness is alloimmunization. Alloimmunization is an immune response in a recipient stimulated by foreign donor cells In the platelet transfusion setting, these responses involve the production of antibodies directed at donor platelet which may lead to inadequate response to platelet transfusion. These alloantibodies may be directed either against platelet specific antigens (HPA) or antigens shared with other tissues (HLA antigens and ABH blood group antigens). Alloimmunization to HLA antigens are more common than to the ABH or HPA antigens. It is caused by transfusion of nonleukodepleted platelet units where the incidence may reach up to 100% [23].
| A. Non-immunologic refractoriness (70-80% cases) 1. Acute bleeding, platelet consumption 2. Fever (any cause) 3. Infection 4. Medications (amphotericin B, vancomycin) 5. Microangiopathies and Disseminated intravascular coagulation (DIC) 6. Splenomegaly B. Immunologic refractoriness (20-30% cases) 1. ABO-mismatched transfusion 2. HLA and/or platelet specific antibodies 3. Autoimmune Thrombocytopenia (Idiopathic Thrombocytopenic Purpura) |
Table 1: Classification of platelet refractoriness.
These challenges are more common in developing countries where there is no practice of universal leukodepletion, gamma irradiation, Nucleic Acid testing for viral makers (HBV, HIV, and HCV), CMV screening and bacterial detection of blood components in most of the blood centers. Management of platelet refractoriness is by first treating the underlying cause. Switching to ABO group specific platelets, preferably collected by apheresis is the next step. If refractoriness still persists then HLA-matched platelets collected by apheresis should be used [25,28]. The serum sample of the patient should be screened for platelet-specific antibodies and using cross-matched platelets should be considered whenever possible.
Post-transplant thrombocytopenia is one of the important prognostic factors influencing transplant related mortality. Earlier studies showed that prolonged thrombocytopenia is a strong negative prognostic indicator of survival in transplant recipients particularly in those cases where thrombocytopenia is related to GVHD. Furthermore thrombocytopenia is correlated with a higher incidence of advanced acute and cGVHD [29]. A retrospective study of 71 patients by Zaja et al. showed that the incidence of mortality was significantly higher in patients with post-SCT thrombocytopenia compared with patients without thrombocytopenia (61.5% vs. 10.5%; p=0.005) [30]. They concluded that post-SCT thrombocytopenia appears to be a strong negative predictor of survival, in which cGVHD seems to be the most frequent pathologic condition associated with thrombocytopenia [30]. Also impaired post-transplant thrombopoiesis is associated with inferior survival [31].
Some of the recent progresses in thrombocytopenia evaluation include assessing MK ploidy and maturation using flow cytometry [13]. Larger studies could be useful in diagnosing and treating the transplant patients with thromobocytopenia. Another recent advance is evaluating levels of SDF-1 and VEGF by ELISA in patients of thromobocytopenia [12]. This would provide good insight into the causes of failure in engraftments of MK. Further study in the assessment of causes for impaired levels of these cytokines and the corrective procedures will be very helpful in treating these patients. Other cytokines that need to be evaluated are stem cell factor, IL-6. Platelet alternatives like recombinant human thrombopoietin (TPO), recombinant IL-6, IL-3, IL-11, amifostine , pegylated thrombopoeitin, lyophilized platelets, fibrinogen-coated albumin microcapsules etc. will be used to manage these patients [24]. However antibodies against recombinant TPO and efficacy of alternatives will remain a major concern.
Thrombocytopenia has remained a major concern in post-HSCT patients. The pathogenesis varies from impaired thrombopoiesis to increased turnover of platelets. It is a strong negative indicator for survival and is often seen in advanced acute and chronic GVHD. The management includes treating the underlying conditions and supportive care with platelet transfusion.