Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 5
Cartilage damage is a major problem in osteoarthritis (OA). To gain insight into the pathogenesis of OA, we have analyzed the differential proteome of articular chondrocytes from these patients. Protein extracts were prepared from cultured chondrocytes from 6 patients with end-stage OA and 6 normal donors and were analyzed by 2D-DIGE. Differentially expressed proteins were identified by mass spectrometry (MS). Significant differential expression was observed for 27 proteins, with 14 underexpressed and 13 overexpressed chondrocyte OA proteins. Of special interest was the identification of destrin, cofilins, gelsolin, annexin A2, glycolytic enzymes, chaperones, cathepsin D, proteasome beta 9 subunit isoform 2 proprotein and proteasome activator hPA28 subunit beta. The altered expression of these proteins is consistent with events such as cytoskeleton binding, protein disruption, apoptosis, and glycolysis, demonstrating the ability of the 2D-DIGE/MS platform to identify proteins with altered expression in chondrocytes from patients with end-stage OA. The identification of these proteins may open new lines of research for this disease.
Keywords: Osteoarthritis, Cartilage, Chondrocytes, Proteomics, 2D-DIGE
2D-DIGE: Two-dimensional Fluorescence Difference Gel Electrophoresis; 2-DE: Two-dimensional Gel Electrophoresis; IEF: Isoelectric Focusing; MALDI-TOF MS: Matrix-assisted Laser Desorption Ionization Time-of-flight Mass Spectrometry; MS: Mass Spectrometry; OA: Osteoarthritis; PFF: Peptide Fragmentation Fingerprint; PMF: Peptide Mass Fingerprint
Osteoarthritis (OA) is a degenerative joint disease. It is the most pervasive form of arthritis and a major cause of disability. Degeneration of articular cartilage in combination with an altered subchondral compartment is a key feature of OA (Ding et al., 2005; Conaghan et al., 2006). In terms of quality of life and chronic disability, OA is second only to cardiovascular diseases (Haq et al., 2003). It is estimated that more than one-third of the population above the age of 35 will experience OA at some point in their lives (McCauley et al., 1998).
Normal articular cartilage is a highly specialized and uniquely designed biomaterial that forms the smooth, gliding surface of diarthrodial joints. It is largely an avascular, aneural and alymphatic matrix that is synthesized and maintained by a sparse population of specialized cells, the chondrocytes. In the adult human, these cells may occupy as little as 2% of the total cartilage tissue volume. The matrix is mainly composed of collagen (mostly type II with lesser amounts of other collagen types) and proteoglycans, principally aggrecan, which is large and aggregates with hyaluronic acid (Aurich et al., 2005). Breakdown of cartilage in OA involves degradation of the extracellular matrix macromolecules and altered expression of chondrocyte proteins necessary for normal joint function (Lane et al., 2000).
Proteomics is an emerging field with widespread potential applications to rheumatic diseases (Ali and Manolios, 2005). A key methodological advance in two-dimensional gel electrophoresis (2-DE) has been the emergence of multiplexing two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) (Unlu et al., 1997). 2D-DIGE directly labels lysine groups on proteins with cyanine CyDye DIGE Fluor minimal dyes before isoelectric focusing (IEF). Protein extract samples are labelled with Cy3 and Cy5 fluorescent dyes, while Cy2 dye is used to label the internal standard, which consists of a pooled sample comprising equal amounts of all samples to be compared. Then the three samples are electrophoresed on a single 2D gel, which allows both direct quantitative comparisons within each gel and the normalization of quantitative abundance values for each protein between gels. The main advantages of 2DDIGE are sensitivity, high reproducibility and wide dynamic range (Unlu et al., 1997; Alban et al., 2003; Righetti et al., 2004; Viswanathan et al., 2006). The combination of 2DDIGE with matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) is a powerful tool for identifying disease-related proteins (Stults and Arnott, 2005).
The aim of this study was to screen proteins with altered expression in chondrocytes from patients with endstage OA as compared to control subjects. The characterization of these proteins should expand knowledge of the pathological processes implicated in the damage of articular cartilage in OA.
Patients and Specimens
Human knee cartilage was obtained during total joint replacement surgery in 6 patients with clinical and radiological features of OA (mean age 72 years, range 61-83), according to the criteria of the American College of Rheumatology (Arnett et al., 1988; Altman et al., 1986). Control samples were obtained from 6 adult donors (mean age 83 years, range 76-91), without a history of joint disease, during orthopaedic surgeries not related to OA. All samples were obtained after patients gave their informed consent, and this study was approved by the local institutional ethics committee.
Isolation and Culture of Chondrocytes
Once cartilage surfaces were rinsed with saline, scalpels were used to cut parallel sections 5 mm apart, vertically from the cartilage surface onto the subchondral bone. These cartilage strips were minced into small pieces (1-3 mm3) and incubated with trypsin at 37ºC for 10 min. After removing the trypsin solution, the tissue cartilage slice was transferred into a digestion buffer containing low-glucose Dulbecco’s modified Eagle’s medium (LG-DMEM; Gibco BRL, Paisley, UK), 5% fetal bovine serum (FBS) (Sigma- Aldrich; St. Louis, MO), antibiotics and 2 mg/ml of clostridial collagenase type IV (Sigma-Aldrich). Tissue was incubated for 12-16 h on a shaker at 37ºC. Thereafter, the cell suspension was collected by centrifugation at 1000 g for 10 minutes and washed 2 times in DMEM, 5% fetal bovine serum (FBS) and antibiotics. Cells were recovered and plated at high density (4 x 106 per 175-cm2 flask; BD) in DMEM supplemented with 10% FBS and antibiotics. Cells were incubated at 37ºC in a humidified gas mixture containing 5% CO2 balanced with air. Chondrocytes were used at 2-3 weeks at confluency in primary culture. Medium changes were performed twice weekly.
Protein Sample Preparation
Trypsinized cells were washed with PBS and centrifuged. Pelleted cells were disrupted with lysis buffer (30 mM Tris-HCl pH 8.5, 7 M urea, 2 M thiourea, 4% CHAPS, 50 mM DTT) and ground with resin and a pestle using the Grinding kit (GE Healthcare). The proteins were precipitated with the 2D Clean-Up Kit (GE Healthcare) and resuspended in lysis buffer without DTT, with pH adjustment to 8.5 at 4°C when necessary. Protein concentration was measured with the RCDC Protein Assay Kit (Bio-Rad).
DIGE Labelling and 2-D Electrophoresis
Protein extract samples from OA patients and donors were labelled with CyDye DIGE Fluor minimal dyes according to the manufacturer’s instructions (GE Healthcare). Briefly, 50 mg of each protein extract was separately labelled at 0ºC in the dark for 30 min with 400 pmol of the Nhydroxysuccinimide esters of cyanine dyes (Cy3 and Cy5 CyDyes) dissolved in 99.8% DMF (Sigma). The internal standard, an equimolecular mixture of all the protein extracts, was labelled with Cy2 dye in the same way. The labelling reaction was quenched by the addition of 1 ml of a 10 mM L-lysine solution (Sigma) and left on ice 10 min. After labelling and quenching, OA patients, controls and internal standard protein samples were mixed adequately (one of each condition per gel) and run in a single gel. A volume of the Rehydration Solution (7 M urea, 2 M thiourea, 4% CHAPS and 0.8% IPG Buffer pH3-11NL) containing 50 mM DTT was added to the mixed samples. Proteins (150μg total protein per gel) were then applied via cup-loading to first dimension immobiline strips (24 cm, IPG Strip pH3- 11NL; GE Healthcare) previously rehydrated in the aforementioned Rehydration Solution containing 100 mM hydroxyethyl disulfide (DeStreak, GE Healthcare). The proteins were separated in the first dimension at 0.05 mA/IPG strip in the IPGphor IEF II System (GE Healthcare) until steady state (42 kVh). Then, the strips were equilibrated and separated by SDS-PAGE on 12% polyacrylamide gels (26 x 20 cm) precast into low fluorescence-glass plates using an Ettan Dalt Six device (GE Healthcare) at 2 W/gel for 1 h and 20 W/gel at 25ºC until the tracking dye had migrated off the bottom of the gel.
Image Acquisition and Analysis
After SDS-PAGE, the gels were scanned on a Typhoon™ 9400 variable mode Imager (GE Healthcare) using appropriate wavelengths and filters for Cy2, Cy3 and Cy5 dyes according to the manufacturer’s protocol. Scans were acquired at 100 mm resolution. After cropping, images were subjected to automated Differential In-gel Analysis (DIA) and Biological Variation Analysis (BVA) using the DeCyder Differential Analysis Software, Release 6.5 (GE Healthcare). After manual inspection of this automatic matching, the statistical module EDA (Extended Data Analysis) provided statistical analysis of samples based on Student’s t-test and multivariate analysis.
In-gel Protein Digestion
A digestion robot (Proteineer DP, Bruker-Daltonics, Bremen, Germany) was employed to reduce, alkylate and digest protein spots of interest excised from the gel following the protocol of (Shevchenko et al., 1996; Shevchenko et al., 2006) with minor variations. Briefly, reduction was performed by incubating the gel plugs with 10 mM dithiothreitol (Amersham Biosciences, Uppsala, Sweden) in 50 mM ammonium bicarbonate (99.5% purity; Sigma Chemical, St. Louis, MO, USA) for 30 min, and 55 mM iodoacetamide (Sigma Chemical) in 50 mM ammonium bicarbonate was used as an alkylating agent for 20 min. The gel pieces were then rinsed with 50 mM ammonium bicarbonate and acetonitrile (gradient grade; Merck, Darmstadt, Germany) and dried under a stream of nitrogen. Modified porcine trypsin (sequencing grade; Promega, Madison, WI, USA), at a final concentration of 8 ng/ml in 50 mM ammonium bicarbonate, was added to the dry gel pieces and the digestion proceeded at 37ºC for 8 h. Finally, the digestion solution was acidified with 0.5% trifluoroacetic acid (99.5% purity; Sigma Chemical) for acid peptide extraction.
Protein Identification by MALDI MS and Database Searching
Each protein digest was mixed 1:1 with 0.5 g/l a-cyano- 4-hydroxycinnamic acid (Bruker-Daltonics) in 33% aqueous acetonitrile and 0.25% trifluoroacetic acid used as a matrix solution, deposited on a prestructured 600 mm MALDI target (Bruker-Daltonics), air-dried, and subjected to peptide mass fingerprint (PMF) and peptide fragmentation fingerprint (PFF) analyses to assess protein identity. Single (MS) and tandem (MS/MS) mass spectra were acquired in an automated analysis loop on an Ultraflex timeof- flight mass spectrometer (Bruker-Daltonics) equipped with a delayed-extraction ion source and a LIFT-MS/MS device (Suckau et al., 2003). Ionisation was achieved by irradiation with a pulsed nitrogen laser operating at 50 Hz and 2 ns pulse width, and 100 to 1000 individual spectra were averaged. To measure fragment ions in the tandem time-of-flight (TOF/TOF) mode, precursor ions were accelerated to 8 kV and selected in a timed ion gate. Fragment ions generated by laser-induced decomposition of the precursor were further accelerated by 19 kV in the LIFT device and their masses were analyzed after passing the ion reflector. Mass data were analysed in an automated fashion using the flexAnalysis software (Bruker-Daltonics). Internal calibration of MALDI-TOF mass spectra was performed using two trypsin autolysis ions with m/z = 842.510 and m/z = 2211.105; for MALDI-MS/MS, calibrations were performed with fragment ion spectra obtained for the proton adducts of a peptide mixture covering the 800-3200 m/ z region. MALDI-MS and MS/MS data were combined through the BioTools program (Bruker-Daltonics) to search a nonredundant protein database (NCBInr; ~5 x 106 entries; National Center for Biotechnology Information, Bethesda, US) using the Mascot software (Matrix Science, London, UK) (Perkins et al., 1999). MALDI-MS and MS/ MS spectra and database search results were manually inspected in detail using the above programs as well as homemade software.
Biological Function
Information about those proteins found to be differentially regulated in chondrocytes from OA patients was retrieved from various proteomics servers (RCSB Protein Data Bank, Babelomics, Expasy Proteomics Server, Information Hyperlinked over Proteins, EBIMed) containing information from the main protein databases (UniProt Knowledgebase, Prosite, IntAct, OMIM and NCBI).
2D-DIGE Analysis of Human Normal and OA Chondrocytes
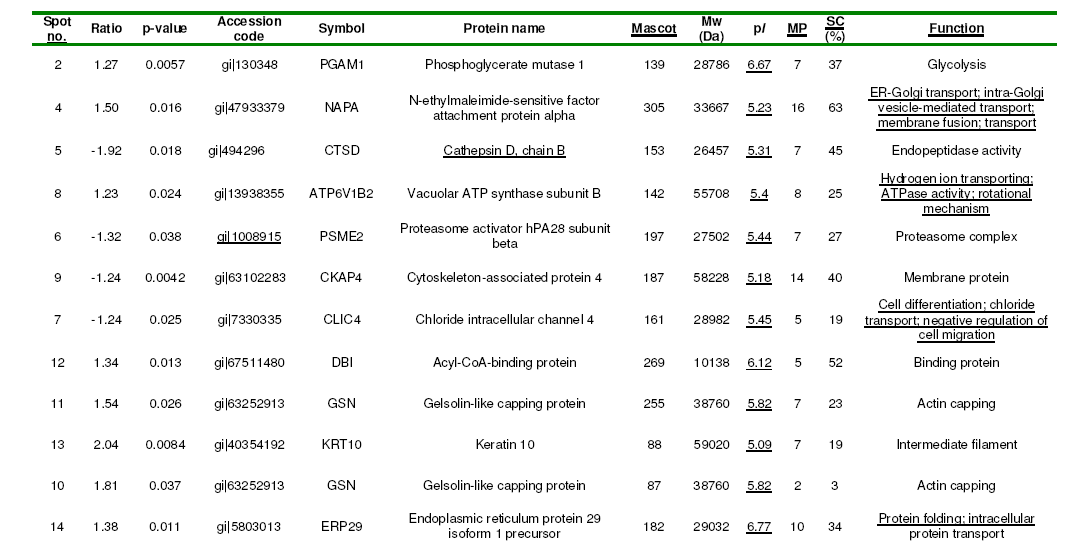
Comparative profiling of protein expression between normal and OA chondrocytes was carried out by 2D-DIGE. Whole-cell lysates (150 mg) from twelve individuals, six OA patients and six donors, were labelled with Cy3 or Cy5 fluorescent dyes. To avoid labelling bias arising from the distinct fluorescence features of gels at different wavelengths, protein extracts were labelled using dye-swapping with either Cy3 or Cy5 fluorescent dyes, so that a given sample type (control or OA) was labelled with both dyes, and then each Cy3/Cy5-labeled sample pair was mixed with a Cy2- labeled internal standard on each gel. After 2-DE, the Cy2, Cy3 and Cy5 channels were individually imaged from each of the gels using mutually exclusive excitation and emission wavelengths. Figure 1 shows a representative differential CyDye staining of chondrocyte proteins, with proteins from one OA sample stained in green (Cy3), proteins from one control sample stained in red (Cy5), the internal standard stained in blue (Cy2) and an overlay of the three dye scanimages. Relative protein quantification across all OA patients and controls was performed using Decyder software. First, the DIA module co-detected the three images of the gel (internal standard and two samples), to provide a consistent and accurate spot ratio measurement in terms of the ratios of the Cy3 and Cy5 spot volumes to the standard Cy2 volume on each gel. Background subtraction, quantification and normalization were automatically applied with low experimental variation. The images individually processed with the DIA module were matched between gels with the BVA module, using the internal standard for gel-to-gel matching. BVA detected the consistency of differences between experimental groups across all the gels, and applied statistics to determine a level of confidence (Student’s t-test) for each of the differences, which were calculated as average ratios for each spot. Finally, the BVA results were loaded on EDA software for additional advanced statistical analysis. The fluorescence volumes of 35 spots showed a significant difference (p<0.05) between normal and OA chondrocytes. From these, 14 spots decreased and 21 spots increased. To identify the proteins, one of the 2-D DIGE-labelled gels was stained with silver (Fig. 2).

Figure 1: 2D-DIGE of chondrocyte proteins. Each individual sample (OA and controls) and a pooled reference sample were labelled with Cy5, Cy3 and Cy2, respectively, mixed, and separated on a 2D-PAGE gel. Gels were scanned and a set of Cy5 (A), Cy3 (B) and Cy2 (C) images were obtained from each gel. An overlay of three dye scan-images was also obtained (D).

Figure 2: 2-D silver-stained gels of chondrocyte proteins. Spots with a Student’s t-test p value less than 0.05 in OA are shown in circles and numbered according to pI and Mw. The circled protein spots were cut out of this gel and subjected to tryptic digestion followed by MS analysis.
Identification of Significantly Altered Expression Spots by MS
Differentially expressed proteins were excised from the silver-stained gel, in-gel digested with trypsin and analysed using MALDI-TOF MS. Combined PMF and PFF analysis followed by interrogating the NCBI database with Mascot enabled the identification of 30 out of 35 (27 proteins) differentially expressed spots in OA chondrocytes (Table 1). Three of the proteins identified were found in more than one spot: Gelsolin-like capping protein (spots 10, 11), Cofilin 1 (non-muscle), isoform CRA_c (spots 15, 26), and Annexin A2 isoform 2 (spots 19, 20). From spot 19, two different proteins, annexin A2 isoform 2 and LIM and SH3 protein 1, were identified.
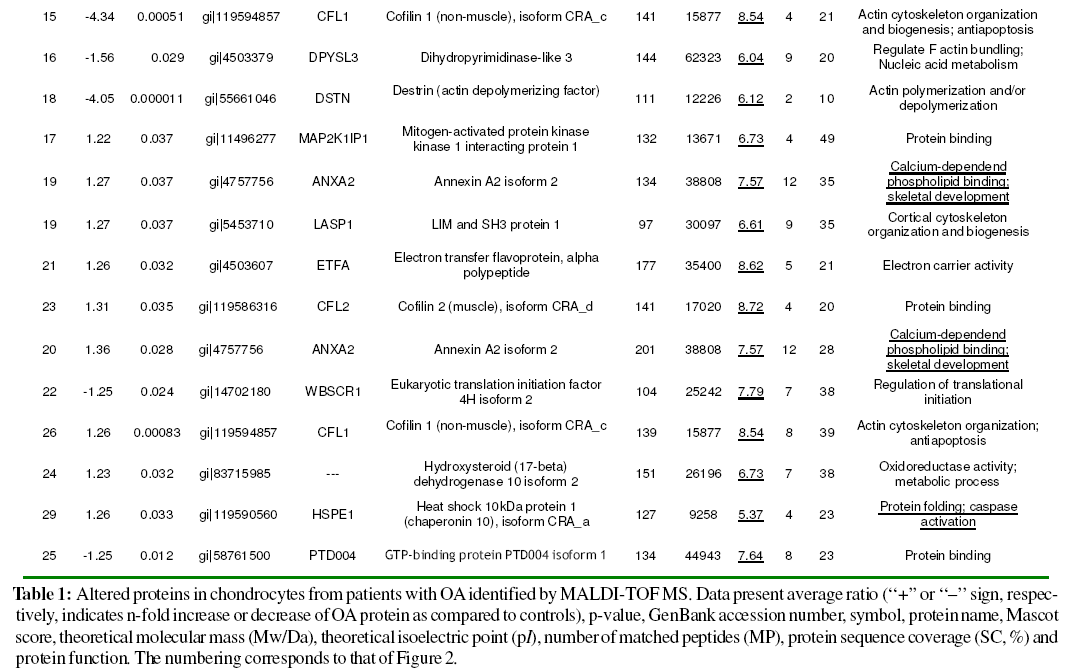
The cellular localization, molecular function and biological process of the identified proteins obtained from the FatiGO ontology database are summarized in Fig. 3. The principal cellular localizations of these 27 proteins were cytoskeleton (22%), mitochondrion (19%), cytosol (15%), endoplasmic reticulum (11%), and proteasome (7%) (Fig. 3A). Protein classification on the basis of molecular function revealed that 21% were cytoskeletal binding proteins. Other proteins detected displayed peptidase (7%), translation initiation factor (4%), proteasome activator (4%), chanel (4%), hydrolase (11%), isomerase (4%), oxidoreductase (11%) and transferase (4%) activities (Fig. 3B). Classification according to biological process showed that a significant proportion of proteins were involved in actin cytoskeleton organization (13%), protein folding (4%), carbohydrate metabolic process (8%), protein metabolic process (4%), regulation of hydrolase activity (8%) and immune response (8%) (Fig. 3C). Table 2 illustrates the functional classification of the main differential proteins discussed in this report.

Figure 3:Gene ontology annotation of the 27 changed proteins identified from human normal and OA chondrocytes. Results were obtained from BABELOMICS Bioinformatics FatiGO tools at http://fatigo.bioinfo.cipf.es/ (Al-Shahrour et al., 2005).
A. Distribution of the identified proteins in various subcellular localizations.
B. Distribution of identified proteins implicated in various molecular functions.
C. Distribution of identified proteins implicated in various biological processes.
Some studies have analysed the secretory profile of normal (Hermansson et al., 2004) and OA (Hermansson et al., 2004; Garcia et al., 2006) articular cartilage by 2-DE and MS. Another study (Ruiz-Romero et al., 2005) has described the proteome of human normal articular chondrocytes in order to establish a novel tool for the study of OA. Very recently, these authors and others (Lambrecht et al., 2007; Ruiz et al., 2008), have examined the proteome of human articular chondrocytes from normal versus OA cartilage using Sypro Ruby stained conventional 2-D gels and image analysis based on PDQuest software.
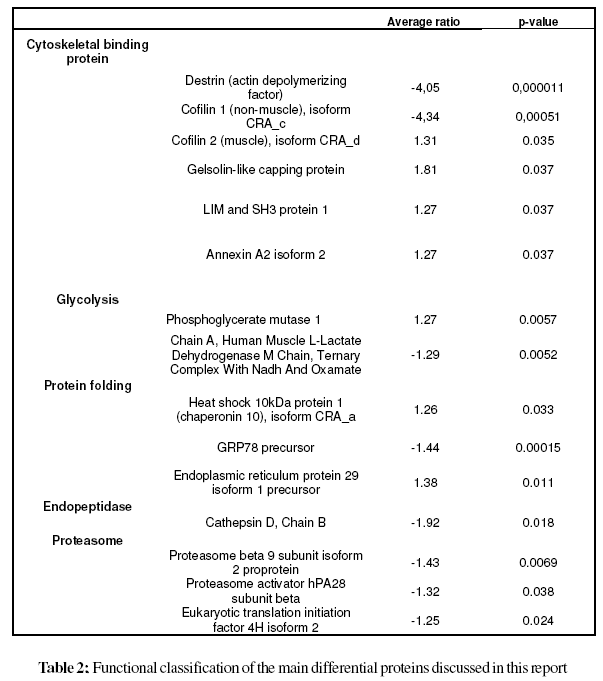
In this study, we have conducted, for the first time, a 2D-DIGE analysis of normal and OA-affected articular chondrocytes in order to identify proteins differentially expressed in OA chondrocytes. The 2D-DIGE technique circumvents many of the issues associated with traditional 2- DE, such as gel-to-gel variation and limited dynamic range. The internal standard sample included on each gel enables accurate comparison of protein abundance between different samples on different gels, and dedicated DeCyder image analysis software facilitates more accurate and sensitive detection and quantitation of changes in protein expression between samples. Nevertheless, a few technical issues need to be considered. First, protein patterns obtained with 2D-DIGE could differ from those revealed by conventional systems. In addition, the fluorescent labelling efficiency of proteins increases with lysine content, which tends to overrepresent proteins with high lysine content. In our study, a significant differential expression pattern was observed for 27 different proteins with 14 decreased and 13 increased chondrocyte proteins.
We identified an elevated number of cytoskeletal binding proteins that displayed significantly changed expression in OA chondrocytes, like destrin (DSTN), cofilins (CFL1, and CFL2), gelsolin (GSN), LIM and SH3 protein 1 (LASP1) and annexin A2 (ANXA2). DSTN belongs to the actin-binding ADF protein family. ADF/cofilins are small proteins that bind to both monomeric and filamentous actin, and their most important physiological function is to increase actin dynamics by depolymerizing filaments from their pointed ends (Paavilainen et al., 2004). Gelsolin-like capping protein is a member of the gelsolin/villin family of actin-regulatory proteins that binds to the free end of the actin filament, thereby preventing further addition of subunits. Some studies have confirmed the importance of actin organization in controlling the chondrocyte phenotype (Loty et al., 1995; Woods, et al., 2005), whilst a gelsolin-like protein has been found upregulated in hypertrophic versus non-hypertrophic chondrocytes (Nurminskaya et al., 1996) The actin cytoskeleton provides the cell with mechanical stability and has been linked to the process of chondrocyte mechanotransduction through which cartilage cells sense and adapt to external mechanical stimuli (Wang et al., 1993; Grodzinsky et al., 2000; Millward-Sadler and Salter, 2004; Guilak, 1995). This process is essential for the functional integrity of articular cartilage through the control of extracellular matrix synthesis and catabolism and the prevention of an inflammatory response to mechanical loading. The cofilin gene, along with its isoform destrin, has been found significantly up-regulated in chondrocytes upon mechanical loading (Campbell et al., 2007). In addition, a high number of actin-remodeling proteins, like cofilin, destrin and gelsolin have been identified in the proteomic map of human normal articular chondrocytes (Ruiz-Romero et al., 2005). Thus, the study of these actin-binding proteins may open new research lines in OA pathogenesis.
ANXA2 belongs to a family of Ca2+ and phospholipidbinding proteins. Previous studies have shown the expression of hypertrophic and terminal differentiation markers, including ANXA2, in OA cartilage (Kirsch et al., 2000). In addition, mineralization and apoptosis were detected in OA cartilage (Kirsch et al., 2000; Hashimoto et al., 1998; Derfus et al., 1996). Wang et al., (2003) hypothesised that up-regulation of annexin gene expression in OA cartilage might lead to annexin-mediated Ca2+ influx into articular chondrocytes and subsequent stimulation of terminal differentiation events in these cells. Terminal differentiation events are required for endochondral bone formation during normal development; however, if these events occur under pathological conditions such as OA, cartilage destruction is triggered. Here, up-regulation of ANXA2 protein expression illustrates the involvement of apoptosis in OA.
Two proteins implicated in glycolysis were found dysregulated in OA chondrocytes: Phosphoglycerate mutase 1 (PGAM1) and lactate dehydrogenase A (LDHA). PGAM1 is involved in the anaerobic enzymatic conversion of glucose to lactate or pyruvate, resulting in energy stored in the form of adenosine triphosphate (ATP), while lactate dehydrogenase A catalyzes the conversion of L-lactate and NAD to pyruvate and NADH in the final step of anaerobic glycolysis. The articular cartilage is an avascular connective tissue with significantly reduced availability of oxygen and glucose as compared to synovial fluid and plasma, and glucose is an important metabolic fuel and structural precursor that plays a key role in the synthesis of extracellular matrix macromolecules in articular cartilage. Chondrocytes survive in an extracellular matrix with limited nutrients and low oxygen tension and, consequently, might sense and respond to low oxygen tension and alterations in extracellular glucose by adjusting cellular metabolism and altering expression of glycolytic enzymes.
We identified two chaperones: Chaperonin 10 (HSPE1) and GRP78. HSPE1, also called heat shock protein 10, is a 70 kDa mitochondrial protein known mainly for its role in intracellular protein folding (Slavotinek and Biesecker, 2001). A role in the modulation of the innate immune response has been proposed for extracellular HSPE1. This molecule has antiinflammatory and immunomodulatory properties via inhibition of downstream events in Toll-like receptor (TLR) activated pathways (Johnson et al., 2005). The treatment of rheumatoid arthritis patients with HSPE1 results in shortterm improvement of disease activity indicators (Vanags et al., 2006). GRP78 is involved in protein folding and assembly in the endoplasmic reticulum (ER). It has also been demonstrated that GRP78 plays a role in the processing and transport of wild type cartilage oligomeric matrix protein (COMP), a large pentameric glycoprotein from the thrombospondin (TSP) group of extracellular proteins, found in the territorial matrix surrounding chondrocytes, in normal chondrocytes and in the retention of mutant COMP in PSACH chondrocytes (Hecht et al., 2001). ERP29, another protein with altered expression in OA chondrocytes, is thought to play a role in the processing of secretory proteins within the ER, possibly by participating in protein folding (Hubbard et al., 2006). The underlying mechanisms by which these folding proteins come to be dysregulated in chondrocytes from OA patients await further study.
Proteases play important roles in tissue damage and destruction in the arthritic joint (Keyszer et al., 1998; Cunnane et al. 2001). The lysosomal and secreted aspartate protease cathepsin D has the potential to contribute to the proteolytic processing of the aggrecan core protein in articular cartilage (Handley et al., 2001). This protein is associated with the process of endochondral ossification and has been proposed to play a role in human bone and cartilage (Nakase et al., 2000). Moreover, cathepsin D has been identified in the proteome of normal human chondrocytes by 2-DE and MS (Ruiz-Romero et al., 2005). Increased levels of matrix degrading enzymes in OA have been involved in the aetiopathogenesis of the disease, including exercise-associated OA. Bowe et al., (2007) found that a high intensity exercise regime not only caused phenotypic changes typical of early OA (Murray et al., 1999) but also an unexpected significant decrease in the percentage of chondrocytes positive for cathepsin D, particularly in cartilage adjacent to the subchondral bone. In agreement with this result, herein a down-regulation of this protease has been detected in OA chondrocytes.
Two proteins related to the proteasome were found downregulated in OA chondrocytes: PSMB9, a large cytosolic protein complex that is responsible for degrading proteins which have been marked for destruction by ubiquitination or by some other means, and proteasome activator hPA28 subunit beta. A detailed analysis of the proteasome in OA cartilage may help to understand the role of these proteases in OA.
The study of the differential intracellular proteomic profile of chondrocytes from patients with end-stage OA by the 2D-DIGE/MS analytical platform has turned out to be an excellent approach to assess altered expression of such proteins as destrin, cofilins, gelsolin, annexin A2, glycolysis enzymes, chaperones, cathepsin D, proteasome beta 9 subunit isoform 2 proprotein and proteasome activator hPA28. Dysregulation of these proteins suggests that several key cellular processes like cytoskeleton binding, protein disruption, apoptosis and glycolysis are altered in OA cartilage, therefore opening up new research lines in OA pathogenesis.
Outlook
Several key cellular processes appear to be altered in OA cartilage, and future work should deal with result validation using conventional (e.g. Western blot) or complementary approaches (DNA microarrays).
Given that diagnosis and prognostication of OA relies on complex criteria, quantitative proteomic approaches will play an increasingly important role in the discovery of improved biomarkers for OA and in understanding OA pathophysiology.
However, the presence of high abundant interfering components in cartilage samples (e.g. aggrecan) that may hide the detection of much lower abundant proteins of interest requires improvement of sample preparation and fractionation. In addition, as the analytical throughput augments, the analysis of more complex samples from larger cohorts of patients will account for intersample variability, an essential issue for the reliable identification of proteome changes in OA cartilage.
Elucidation of effective strategies for therapy, with emphasis on mechanisms that affect the function of chondrocytes and interactions with surrounding tissues are currently in progress including biologic therapies targeting specific proteins. In this sense, our results indicate that cytoskeletal binding proteins, glycolysis-related proteins, proteases and proteins related to apoptosis and proteasome are candidates to therapeutic strategies in OA.
We wish to thank the orthopaedic surgeons at the Hospital Clínico San Carlos for providing articular cartilage samples. This work was supported by the grants FMMA and FIS 04/1698. Raquel Rollin holds a research contract from the “Fundación para la Investigación Biomédica-Hospital Clínico San Carlos Madrid.”