Journal of Horticulture
Open Access
ISSN: 2376-0354
ISSN: 2376-0354
Research Article - (2014) Volume 1, Issue 2
Guava (Psidium guajava L.) is one of the important fruit crops which are grown in tropical and subtropical countries including India. Wilt is an important disease in guava which is caused by Fusarium oxysporum f. sp. psidii (Fop) as a major obstacle for guava fruit production. It is manifested symptomatically with alterations in the development process such as premature shedding of leaves, pre-maturation of fruits, entire/whole tree defoliation and eventually death of the plant. In this study a colony PCR assay was developed for direct amplification of Internal Transcribed Spacer (ITS) region of fungal rDNA of Fop isolates. For this, two sets of primers were developed for amplification of ITS region. . The sensitivity of both the primer pair was ranged up to 10-6 and 10-7 dilutions of the pure mycelium suspension. This study provides new insight for rapid, sensitive and specific molecular detection of Fop isolates, and also useful for earlier diagnostic for better management of guava wilt disease.
<Keywords: Fusarium oxysporum f. sp. psidii; Diagnostic; ITS-PCR; Molecular detection; Wilt disease
Guava (Psidium guajava L.), an arborescent shrub or a small tree, is one of the very productive and highly profitable fruit crop. It belongs to the family myrtaceae and is being grown all over the sub-tropical and tropical regions globally. It contains high dietary value and good flavor. Wilt of guava is one of the most important diseases of guava, especially in India. In earlier studies, a number of reports are available on wilt disease is caused by Fusarium species [1-4] but the most common fungi associated with wilt disease is F. oxysporum f. sp. psidii [5,6]. However, a need arise to identify F. oxysporum from guava wilt disease. The morphological and cultural characteristics are not sufficient for pathogen characterization as these are easily influenced by environmental factors and unable to emphasize genetic information of isolates [7,8]. Polymerase Chain Reaction (PCR) has been widely assessed in the field of mycology for diverse genetic analyses, specific detection, phylogenetic studies for identification and taxonomy studies [9]. These molecular studies require a number of laborious steps, including preparation of a DNA template by extraction of DNA from fungi [10]. Recently, Roberto et al. [11] reported that the omission of the DNA extraction procedure, which significantly decreases time and cost; and also avoids the risk of contamination during the DNA extraction process.
Therefore, Polymerase Chain Reaction (PCR) assay has been employed for molecular diagnostic of any pathogens [12,13]. However, the methods currently used more are often based on the analysis of ribosomal RNA (rRNA) gene (or rDNA) sequences that are universal and contain both conserved and variable regions, allowing discrimination at different taxonomic levels [7]. Thus, ribosomal RNA (rRNA) consists (18S-ITS1-5.8S-ITS2-28S) is widely used for molecular identification and phylogenetic analysis. It is the RNA component of the ribosome, the cell structure which provides the site of protein synthesis in all cells. It is present in all prokaryote and eukaryote that can be targeted for amplification to identify the pathogen [7]. In contrast, molecular approaches have been developed for fungal systematic studies, including RAPD analysis [14], specific diagnostic PCR primers [15]. Since these molecular studies require a number of laborious steps, including preparation of a DNA template by extraction of DNA from fungi [10]. Recently, culture-independent PCR assay has been developed for molecular detection of F. oxysporum from soil samples [16]. To avoid the time, labour, cost of PCR assay and also risk of contamination during genomic DNA preparation, a direct colony PCR for fungal cultures could be an ideal approach for early diagnosis of F. oxysporum. The aim of present study is to develop a colony PCR assay to amplify ITS region of F. oxysporum isolated from guava wilt disease.
Isolation and maintenance of Fusarium isolates
For the development of colony PCR assay, previously identified Fusarium isolates from guava wilt samples were used in this study (Table 1). The fungal isolates were deposited at National Agriculturally Important Microbial Culture Collection (NAIMCC), Mau, India and also maintained in Potato Dextrose Agar (PDA) medium at 4°C in Molecular Diagnostics Laboratory, Central Institute for Subtropical Horticulture, Lucknow, India.
| Culture ID | Isolate | Geographic origin | Colony colour | Culture accession No. | GenBank accession No. |
|---|---|---|---|---|---|
| Fop-30 | F. oxysporum | Unno (U.P.) | Light brown | (NAIMCC-F-01998) | KC357563 |
| Fop-44 | F. oxysporum | Farukhabad (U.P.) | Light pink | (NAIMCC-F-02084) | KC292502 |
| Fop-48 | F. oxysporum | Farukhabad (U.P.) | Pink | (NAIMCC-F-00813) | KC357565 |
| Fop-51 | F. oxysporum | Shamsabad (U.P.) | Dark yellow | (NAIMCC-F-02000) | KC357562 |
| Fs-206 | F. solani | Gopalganj (Bihar) | Pink | - | - |
| Fop-84 | F. oxysporum | Kanpur (U.P.) | Light brown | (NAIMCC-F-00815) | KC357561 |
Table 1: Details of isolates of Fusarium spp. along with their geographic location used in present study.
Preparation of template for PCR assay
The purified and identified F. oxysporum isolates were grown on PDA medium at 28 ± 2°C for 5 days. Small amount of mycelia was scratched with a micropipette tip and suspended in 50 μl of 1X Tris-EDTA (pH 8.0) in 1.5 ml fresh eppendorf tube. It was vortexed thoroughly and also centrifuged at 10,000 rpm for 10 seconds and 1 μl of supernatant was used as template for PCR assay.
Design and validation of new species-specific primers
A set of new species-specific primers were designed by using PRIMER3 tools (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3.cgi) based on ITS1-5.8S-ITS2 sequence of reference isolate of F. oxysporum f. sp. psidii-44 (Fop-44). The reference sequence of Fop-44 was submitted to the GenBank and assigned under accession number KC292502 (Figure 1). The specificity of the primers was also validated by BLAST for searching the primer sequences (http://www.ncbi.nlm.nih.gov/BLAST). The list of primers for F. oxysporum used in this study was given in Table 2.

Figure 1: BLAST result of representative isolate Fop-44 (NAIMCCF- 2084).
| Primer | Sequence (5′→3′) | Expected product size (bp) | References |
|---|---|---|---|
| ITS1 ITS4 |
TCCGTAGGTGAACCTGCG G TCCTCCGCTTATTGATATGC |
570 | [17] |
| ITS1F ITS1R | CCAGAGGACCCCCTAACTCT GCCTGAGGGTTGTAATGACG |
230 | [18] |
| 172F 447R |
GATCTCTTGGCTCTGGCATC CTCTCCAGTTGCGAGGTGTT |
280 | This study |
Table 2: List of primers used in present study.
PCR assay conditions
PCR reaction is performed in a thermal cycler (Eppendorf). The reaction mixture 25 μl consisted of 1 μl of mycelium supernatant suspension, 1X PCR buffer, 1. 25 U Taq DNA polymerase (Fermentas), 2 mM MgCl2, 0.2 mM dNTPs and 0.5 μM each of the primers. We used primers ITS1 and ITS4 for amplification of ITS region [17].
While, the rapid detection of Fusarium species was performed using specific primers ITS1F and ITS1R [16]. Thus, we have designed and synthesized a set of new primers (172F/447R) for detection of F. oxysporum f. sp. psidii, isolated from guava wilt disease. A concentration of 1 × 105 - 1 × 106 spore per PCR reaction was used in our experiments as a DNA template. The PCR reaction condition was followed as initial denaturation at 94°C for 3 min, subsequently by 29 cycles of denaturation 94°C for 1 min, annealing temperature of primers at 53°C (ITS1F/ITS1R) and extension at 72°C for 1 min. A final extension at 72°C for 5 min was used. While other PCR condition perform was similar except annealing temperature at 54.5°C (172F/447R). The 10 μl of PCR product was analyzed on 1.2% agarose gel with ethidium bromide at 8V/cm and also visualized under UViPro Transilluminator.
Whereas, nested PCR included the two rounds amplification using universal primers (ITS1/ITS4) for first round and two set of ITS based internal Fusarium species specific primers (ITS1F/ITS1R) and F. oxysporum f. sp. psidii specific primers (172F/447R) was used for second round. Thus, PCR was performed in 25 μl of PCR mixture, including 1 μl of PCR product of first round of PCR using ITS1 and ITS4 primers with two different set of primers 0.5 μM ITS1F/ITS1R and 0.5 μM of 172F/447R, a 0.5 μl mixture containing 2.5 μM of each dNTP and 1.25 U Taq DNA polymerase (Fermentas). The PCR program was followed at 94°C for 3 min, 30 cycles of 94°C for 1 min, 53°C (ITS1F/ITS1R) and 54.5°C (172F/447R) for 1 min, and 72°C for 1 min while the final extension at 72°C for 5 min.
Sensitivity of PCR assay
To determine the sensitivity PCR assay, we tested a dilution series of F. oxysporum f. sp. psidii isolates. We tested dilution series of spores ranging from 10-1 to 10-7. These diluted samples were used as template for PCR amplification using a set of primers (ITS1F-ITS1R and 172F-447R) with above PCR conditions.
Analysis of ITS region
Guava wilt is a serious disease affected the parts of plant such as stems, twigs, leaves, roots and fruits. The causal organism has been identified as F. oxysporum f. sp. psidii. In the present study, we developed a colony PCR for rapid, sensitive and cost effective molecular diagnostic of guava wilt pathogen, F. oxysporum. This PCR assay could be useful for reducing the time, cost and laboratory practice. There is also risk of other pathogen contamination during the extraction of genomic DNA for PCR assay [10,18,19]. We focused on the ITS regions of ribosomal genes (Figure 2) for the construction of primers that can be used to identify Fusarium spp. In this study, we selected ITS1-5.8S-ITS2 region for amplification using previously reported primer (ITS1/ITS4) and we obtained approximately 570 bp common fragment in all isolates. This indicates that there was no size variation in ITS+5.8S rDNA region among Fusarium isolates. As shown in Figure 3, expected amplicon of PCR product was obtained without any PCR inhibitor.
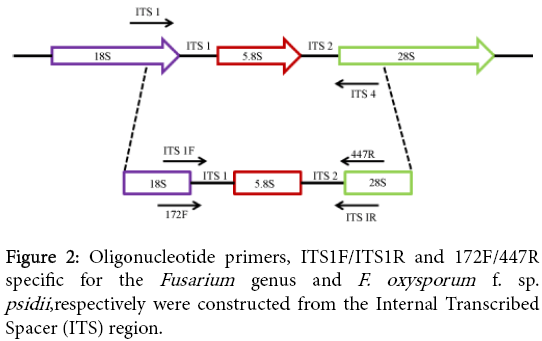
Figure 2: Oligonucleotide primers, ITS1F/ITS1R and 172F/447R specific for the Fusarium genus and F. oxysporum f. sp. psidii ,respectively were constructed from the Internal Transcribed Spacer (ITS) region.
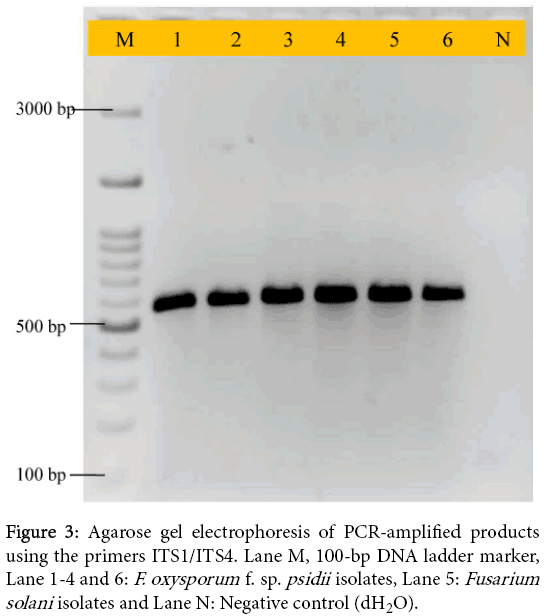
Figure 3: Agarose gel electrophoresis of PCR-amplified products using the primers ITS1/ITS4. Lane M, 100-bp DNA ladder marker, Lane 1-4 and 6: F. oxysporum f. sp. psidii isolates, Lane 5: Fusarium solani isolates and Lane N: Negative control (dH2O).
To detect Fusarium species and also F. oxysporum, a set of primer pairs (ITS1F/ITS1R and newly developed primers: 172F/447R) were used for amplification of conserved region of ITS. We successfully obtained expected size of amplicon in both 230 bp for Fusarium species and 280 bp for Fop isolates. Therefore, isolate Fs-206 was isolated from Gopalganj (Bihar) which was amplified using primers (ITS1F/ITS1R), while no amplification was obtained using primers (172F/447R). This isolate may be other species of Fusarium. The expected size of amplicon was shown in Figure 4A and Figure 4B. In earlier report, PCR assays have been developed for disease diagnosis, monitoring and forecasting programs [20]. A PCR assay has been developed based on amplification of ITS-5.8S rDNA using a set of primers such as ITS1F-ITS1R [16].
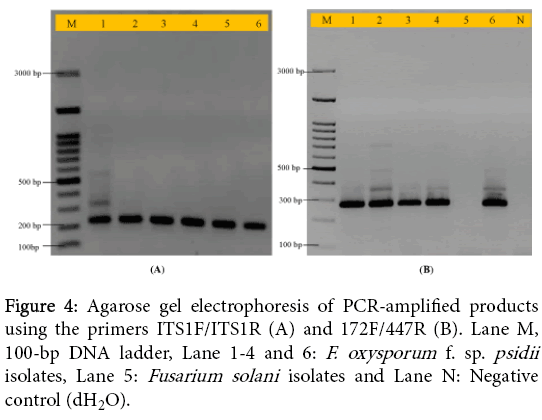
Figure 4: Agarose gel electrophoresis of PCR-amplified products using the primers ITS1F/ITS1R (A) and 172F/447R (B). Lane M, 100-bp DNA ladder, Lane 1-4 and 6: F. oxysporum f. sp. psidii isolates, Lane 5: Fusarium solani isolates and Lane N: Negative control (dH2O).
These previously developed primers were used in present study for rapid detection of Fusarium species. Whereas, the effectiveness of PCR assay for detection and discrimination of F. oxysporum causing wilt disease in guava was demonstrated. The newly designed set of primers gave 280 bp of amplicon fragment in all isolates of Fop which could be able to earlier diagnostic of guava wilt disease.
Nested PCR amplification and analysis
In the present study, a nested PCR was developed to amplify the variable region of ITS of all F. oxysporum which infect wilt disease in guava. Nested PCR uses two sets of primers, one set is external region and second set is internal region amplification of ITS region. The first round of PCR amplification was carried out using ITS1/ITS4 primers and for second round was done using primers (ITS1F/ITS1R). Two new specific primers were evaluated; internal to the nucleotide sequence flanked by universal primers ITS1/ITS4. The specificity and sensitivity of the primer sets ITS1F/ITSR and 172F/447R was shown by amplification of 230 and 280 bp products from second round of PCR amplification in all the isolates of Fusarium spp. (Figure 5A and 5B). Also in nested PCR no amplification was obtained in isolate Fs-206 with primers 172F/447R.
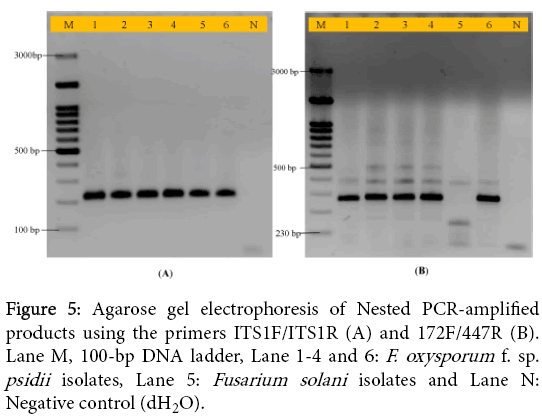
Figure 5: Agarose gel electrophoresis of Nested PCR-amplified products using the primers ITS1F/ITS1R (A) and 172F/447R (B). Lane M, 100-bp DNA ladder, Lane 1-4 and 6: F. oxysporum f. sp. psidii isolates, Lane 5: Fusarium solani isolates and Lane N: Negative control (dH2O).
In the present study, the sensitivity of detection of fungal pathogens can be appreciably enhanced by nested PCR. The conventional nested PCR reaction was shown by amplification of 230 and 280 bp products from Fusarium suspension of all isolates. It is a powerful tool for sensitive and specific molecular diagnostic of F. oxysporum. In earlier studies, nested PCR showed that microsatellite regions could be used for developing highly sensitive PCR detection systems for other fungal pathogens infecting various crops [21]. As the isolation based methods are time consuming, a nested PCR was developed to accelerate and simplify the process of detection was designed based on the genome-specific sequence of Fusarium.
Sensitivity and specificity of nested PCR assay
To determine the sensitivity and specificity of the direct colony PCR, a serial dilution of reference fungal isolate from corresponding species were tested. Figure 6A and Figure 6B showed the results of agarose gel electrophoresis of representative PCR products of the serial dilution of the fungal strains using a set of primers ITS1F/ITS1R and 172F/447R, and the sensitivity of each set of the tests ranged up to 10-6 and 10-7 dilutions, respectively. It could be used for amplification of F. oxysporum isolates directly from mycelium suspension. The sensitivity of the detection method has to be at high levels in order to detect fungal pathogens, especially those causing wilts. We can be further used developed nested PCR for specific detection of not only F. oxysporum but also other guava pathogens or similar approach can be applied for other pathogens.
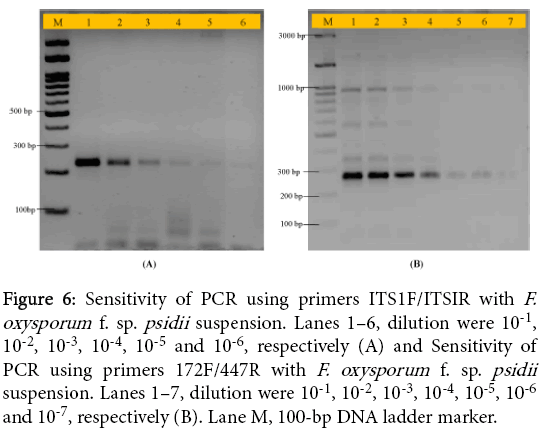
Figure 6: Sensitivity of PCR using primers ITS1F/ITSIR with F. oxysporum f. sp. psidii suspension. Lanes 1–6, dilution were 10-1, 10-2, 10-3, 10-4, 10-5 and 10-6, respectively (A) and Sensitivity of PCR using primers 172F/447R with F. oxysporum f. sp. psidii suspension. Lanes 1–7, dilution were 10-1, 10-2, 10-3, 10-4, 10-5, 10-6 and 10-7, respectively (B). Lane M, 100-bp DNA ladder marker.
In the previous report suggested that the possibility of direct PCR from hyphae of a limited number of mold species [19,21]. Although, several simplification of DNA extraction protocols have been reported, most of them involve the mechanical disruption of mycelia, use of organic solvents, microwave irradiation and various centrifugation steps. The most of process is time-consuming usually DNA extraction and can hinder its effective deployment when either a large number samples need to be analyzed [22], disrupting the cell and need excessive amounts of material [23]. This approach was adopted for detection of Fusarium species that are difficult to identify based on the morphological characteristics. The developed nested PCR assay facilitates the detection and confirmation of Fusarium species and applied during pathogen control activities. The nested PCR assay is a rapid and low-cost, and efficient for the identification and discrimination of genus F. oxysporum f. sp. psidii.
In conclusion, a wide range of diagnostic techniques have been applied for detection, identification and quantification of fungal pathogens which are present in the infected plants, propagative plant materials and postharvest. This assay will be helpful in detection of pathogens directly from infected plant leaf or tissue to avoid DNA extraction protocol. However, time, specificity, sensitivity and cost-effectiveness are major challenge that may determine the suitability and choice of the diagnostic assay. Our newly designed nested PCR based assay could be useful for earlier, rapid, specific and sensitive molecular diagnostic of F. oxysporum f. sp. psidii that may be helpful in management of guava wilt.
We thank to Director, CISH, Lucknow and Head of Crop Protection Division, CISH, Lucknow for their necessary facilities. We gratefully acknowledged to Vice Chancellor, Integral University for encouragement and support.