Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2014) Volume 3, Issue 1
A series of novel polar benzimidazoles were synthesized in a 4-step reaction starting from basic compound 4-fluoro-3-nitrobenzoic acid in good to excellent yield. The compounds generally fulfill the Lipinski’s rule of five to show their potential as drug lead compounds. The compounds were screened for their acetylcholinesterase (AChE) inhibitory activities and the most potent compound was found to be 5e which gave IC50 value of 31.04 μM. The structure of the novel polar benzimidazoles were characterized and confirmed by elemental and mass spectral analyses as well as 1H and 13C NMR spectroscopic data. All the compounds were found to be non-toxic when tested with VERO cells at 50 μM.
<Substituted benzimidazoles are an important class of heterocycles that exhibit a broad spectrum of pharmacological properties, for it assumes the position of a privileged structure in drug discovery research. Derivatives of 1,2-substituted benzimidazoles have been reported as antagonists [1] against prostaglandin D2 and angiotensin II receptors [2]. Similarly, substituted benzimidazoles have been patented as dopamine-β-hydroxylase inhibitors [3]. Hori et al. [4] reported several modified benzimidazoles, serving as guanine biomimetics that selectively inhibit endothelial cell growth and suppress angiogenesis in vitro and in vivo. In nature, the benzimidazole nucleus constitutes an important part of the vitamin B12 structure.
In continuation of our efforts in drug design [5], and especially in the area of Alzheimer’s disease [6], we became interested in polar benzimidazoles as they are more water soluble and potentially less toxic to human [7]. To increase the water solubility we will have to incorporate functionally active polar groups in the structure, and in this case, an ethyl piperidine group at position-2 of the benzimidazole core. It was shown that a methyl piperidine substituent was able to increase water solubility of a compound by 2 orders of magnitude [8]. Since benzimidazole derivatives have been widely used in other areas such as anti-cancer [9] and anti-mycobacterial [10] agents, their pharmacokinetics are well understood. Apart from that, there is also a recent report of benzimidazole having potent anti-tumor activity [11]. In view of the diverse biological applications of benzimidazoles, they represent a good lead in developing new drugs.
All chemicals were supplied by Sigma-Aldrich (USA) and Merck Chemicals (Germany). Purity of the compounds was checked on thin layer chromatography (TLC) plates (silica gel G) in the solvent system chloroform-methanol (9:1). The spots were located under short (254 nm)/long (365 nm) UV light. Elemental analyses were performed on Perkin Elmer 2400 Series II CHN Elemental Analyzer and were within ± 0.4% of the calculated values. 1H and 13C NMR were performed on Bruker Avance 300 (1H: 300 MHz, 13C: 75 MHz) spectrometer in CDCl3 using TMS as internal standard. Direct-infusion mass spectra were recorded on Varian 320-MS TQ LC/MS using ESI.
Preparation of Ethyl-4-fluoro-3-nitrobenzoate (1)
4-Fluoro-3-nitrobenzoic acid (5 g, 27 mmol) was refluxed in ethanol (50 mL) and concentrated H2SO4 (2 mL) for 8 hours. After completion of reaction (as evident from TLC), the solvent was evaporated under reduced pressure. The aqueous layer was extracted with ethyl acetate (25 mL×3). The organic layer was dried over Na2SO4 and concentrated under reduced pressure to yield 1 as cream-colored powder (75%).
Preparation of N-(3-aminopropyl)imidazole (2)
N-(2-aminoethyl) piperazine (1.30 mL, 9.90 mmol) and N,NDiisopropylethylamine, DIPEA (0.49 mL, 2.78 mmol) were mixed in dichloromethane (10 mL). Ethyl-4-fluoro-3-nitrobenzoate, 1 (0.5 g, 2.34 mmol) was added very slowly over 5 minutes. The reaction mixture was stirred overnight at room temperature. The reaction mixture was then washed with water (10 mL×2) followed by 10% Na2CO3 solution (10 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure to afford 2 as yellow solid (92%).
Preparation of Ethyl 4-(2-(piperazin-1-yl)ethylamino)-3- aminobenzoate (3)
N-(3-aminopropyl)imidazole, 2 (0.322 g, 1 mmol), ammonium formate (0.189 g, 3 mmol) and Pd/C (50 mg) were mixed in ethanol (10 mL). The reaction mixture was refluxed until completion (solution turned colorless). The reaction mixture was then filtered through Celite 545. The filtrate was evaporated under reduced pressure. It was resuspended in ethyl acetate and washed with water, dried over Na2SO4 and evaporated to dryness to yield 3 (85%) which was used without further purification.
General procedure for the preparation of sodium bisulfite addcuts of 4-substituted benzaldehyde (4a-e)
Appropriate benzaldehyde (10 mmol) was dissolved in ethanol (20 mL). Sodium metabisulfite (15 mmol) in 5 mL water was added in portion over 5 minutes. The reaction mixture was stirred at room temperature for 1 hour and subsequently stirred at 4°C overnight. The precipitate formed was filtered and dried to afford sodium bisulfite adducts (96%).
General procedure for the preparation of 2-substituted benzimidazole derivatives (5a-5e)
Ethyl 4-(2-(piperazin-1-yl)ethylamino)-3-aminobenzoate, 3 (1 mmol) and various sodium bisulfite adducts, 4a-e (1.5 mmol) were dissolved in DMF (5 mL). The reaction mixture was stirred at 90°C under N2 atmosphere for 24-48 hours. After completion of reaction (evident by TLC), the reaction mixture was diluted in ethyl acetate (25 mL) and washed with water (10 mL×3). The organic layer was collected, dried over Na2SO4 and evaporated under reduced pressure to afford compounds 5a-5e in 77-89% yields.
Ethyl 2-phenyl-1-(2-(piperazin-1-yl)ethyl)-1H-benzo[d] imidazole-5-carboxylate (5a)
This compound was obtained as yellow oil. Yield: 87%. 1H NMR (CDCl3, 300 MHz): δH=1.43 (t, 3H, J=7.2 Hz); 2.24 (t, 4H, J=4.8 Hz); 2.78 (t, 2H, J=6.9 Hz); 3.11 (t, 4H, J=4.8 Hz); 3.50 (t, 2H, J=6.9 Hz); 4.35 (q, 2H, J=7.2 Hz); 7.20-7.80 (m, 6H); 8.05 (dd, 1H, J1=8.4 Hz, J2=1.5 Hz); 8.55 (s, 1H) ppm. 13C NMR (CDCl3, 75 MHz): δC=14.38, 42.79, 53.90, 55.49, 60.93, 62.01, 109.72, 122.44, 124.57, 124.73, 125.36, 129.15, 131.00, 132.08, 138.77, 142.73, 154.62, 167.00 ppm. LC-MS ESIMS: m/z 380.3 [M+H]+. Anal Calc for C22H25N3O3: C, 69.82%; H, 6.92%; N, 14.80%. Found: C, 69.62%; H, 6.97%; N, 14.95%.
Ethyl 2-(4-hydroxyphenyl)-1-(2-(piperazin-1-yl)ethyl)-1Hbenzo[ d]imidazole-5-carboxylate (5b)
This compound was obtained as light brown powder. Yield: 89%. 1H NMR (CDCl3, 300 MHz): δH=1.44 (t, 3H, J=7.2 Hz); 2.21 (t, 4H, J=4.8 Hz); 2.76 (t, 2H, J=6.9 Hz); 3.10 (t, 4H, J=4.8 Hz); 3.48 (t, 2H, J=6.9 Hz); 4.34 (q, 2H, J=7.2 Hz); 6.85 (d, 2H, J=8.4 Hz); 7.39 (d, 2H, J=8.4 Hz); 7.87 (d, 1H, J=8.4 Hz); 8.01 (dd, IH, J1=8.4 Hz, J2=1.5 Hz); 8.54 (s, 1H) ppm. 13C NMR (CDCl3, 75 MHz): δC=14.38, 24.54, 42.79, 51.76, 53.85, 57.50, 61.12, 109.97, 122.49, 125.57, 125.82, 127.35, 127.76, 129.63, 130.03, 138.48, 142.50, 154.19, 167.05 ppm. LC-MS ESIMS: m/z 396.3 [M+H]+. Anal Calc for C22H25N3O3: C, 66.99%; H, 6.64%; N, 14.20%. Found: C, 66.75%; H, 6.84%; N, 14.33%.
Ethyl 1-(2-(piperazin-1-yl)ethyl)-2-p-tolyl-1H-benzo[d] imidazole-5-carboxylate (5c)
This compound was obtained as brown oil. Yield: 77%. 1H NMR (CDCl3, 300 MHz): δH=1.44 (t, 3H, J=7.2 Hz); 2.23 (t, 4H, J=4.8 Hz); 2.45 (s, 3H); 2.77 (t, 2H, J=6.9 Hz); 3.11 (t, 4H, J=4.8 Hz); 3.49 (t, 2H, J=6.9 Hz); 4.36 (q, 2H, J=7.2 Hz); 6.87 (d, 2H, J=8.4 Hz); 7.39 (d, 2H, J=8.4 Hz); 7.88 (d, 1H, J=8.4 Hz); 8.01 (dd, 1H, J1= 8.4 Hz, J2=1.5 Hz); 8.55 (s, 1H) ppm. 13C NMR (CDCl3, 75 MHz): δC=14.38, 42.80, 51.75, 53.90, 57.54, 61.12, 109.97, 116.49, 118.73, 122.49, 124.30, 126.41, 128.50, 129.63, 130.06, 138.49, 142.48, 154.17, 159.07, 167.00 ppm. LCMS ESI-MS: m/z 394.3 [M+H]+. Anal Calc for C22H25N3O3: C, 70.38%; H, 7.19%; N, 14.27%. Found: C, 70.08%; H, 7.40%; N, 14.26%.
Ethyl 2-(4-methoxyphenyl)-1-(2-(piperazin-1-yl)ethyl)-1Hbenzo[ d]imidazole-5-carboxylate (5d)
This compound was obtained as light brown powder. Yield: 86%. 1H NMR (CDCl3, 300 MHz): δH=1.43 (t, 3H, J=7.2 Hz); 2.21 (t, 4H, J=4.8 Hz); 2.75 (t, 2H, J=6.9 Hz); 3.09 (t, 4H, J=4.8 Hz); 3.48 (t, 2H, J=6.9 Hz); 3.87 (s, 3H); 4.36 (q, 2H, J=7.2 Hz); 6.86 (d, 2H, J=8.4 Hz); 7.37 (d, 2H, J=8.4 Hz); 7.80 (d, 1H, J=8.4 Hz); 8.00 (dd, 1H, J1=8.4 Hz, J2=1.5 Hz); 8.53 (s, 1H) ppm. 13C NMR (CDCl3, 75 MHz): δC=14.39, 42.81, 51.75, 53.90, 56.19, 57.54, 61.12, 110.04, 116.52, 118.73, 122.49, 124.30, 126.41, 128.50, 129.65, 130.06, 138.49, 142.48, 154.16, 159.33, 167.02 ppm. LC-MS ESI-MS: m/z 410.3 [M+H]+. Anal Calc for C22H25N3O3: C, 67.63%; H, 6.91%; N, 13.72%. Found: C, 67.50%; H, 7.02%; N, 13.86%.
Ethyl 2-(4-(trifluoromethyl)phenyl)-1-(2-(piperazin-1-yl) ethyl)-1H-benzo[d]imidazole-5-carboxylate (5e)
This compound was obtained as yellow oil. Yield: 85%. 1H NMR (CDCl3, 300 MHz): δH=1.43 (t, 3H, J=7.2 Hz); 2.31 (t, 4H, J=4.8 Hz); 2.73 (t, 2H, J=6.9 Hz); 2.95 (t, 4H, J=4.8 Hz); 4.40 (q, 2H, J=7.2 Hz); 4.41 (t, 2H, J=6.9 Hz); 7.48 (d, 1H, J=8.4 Hz); 7.82 (d, 2H, J=8.4 Hz), 7.97 (d, 2H, J=8.4 Hz), 8.09 (dd, 1H, J1=8.4 Hz, J2=1.5 Hz); 8.55 (s, 1H) ppm. 13C NMR (CDCl3, 75 MHz): δC=14.39, 42.76, 51.75, 53.87, 57.48, 61.10, 109.97, 122.49, 124.91, 125.57, 125.82, 125.85, 125.88, 129.73, 130.00, 138.68, 142.50, 154.16, 167.03 ppm. LC-MS ESI-MS: m/z 448.2 [M+H]+. Anal Calc for C23H25N4O2F3: C, 61.87%; H, 5.64%; N, 12.55%. Found: C, 61.75%; H, 5.38%; N, 12.79%.
The sequence for the formation of the novel benzimidazole derivatives is proposed and summarized in Scheme 1.
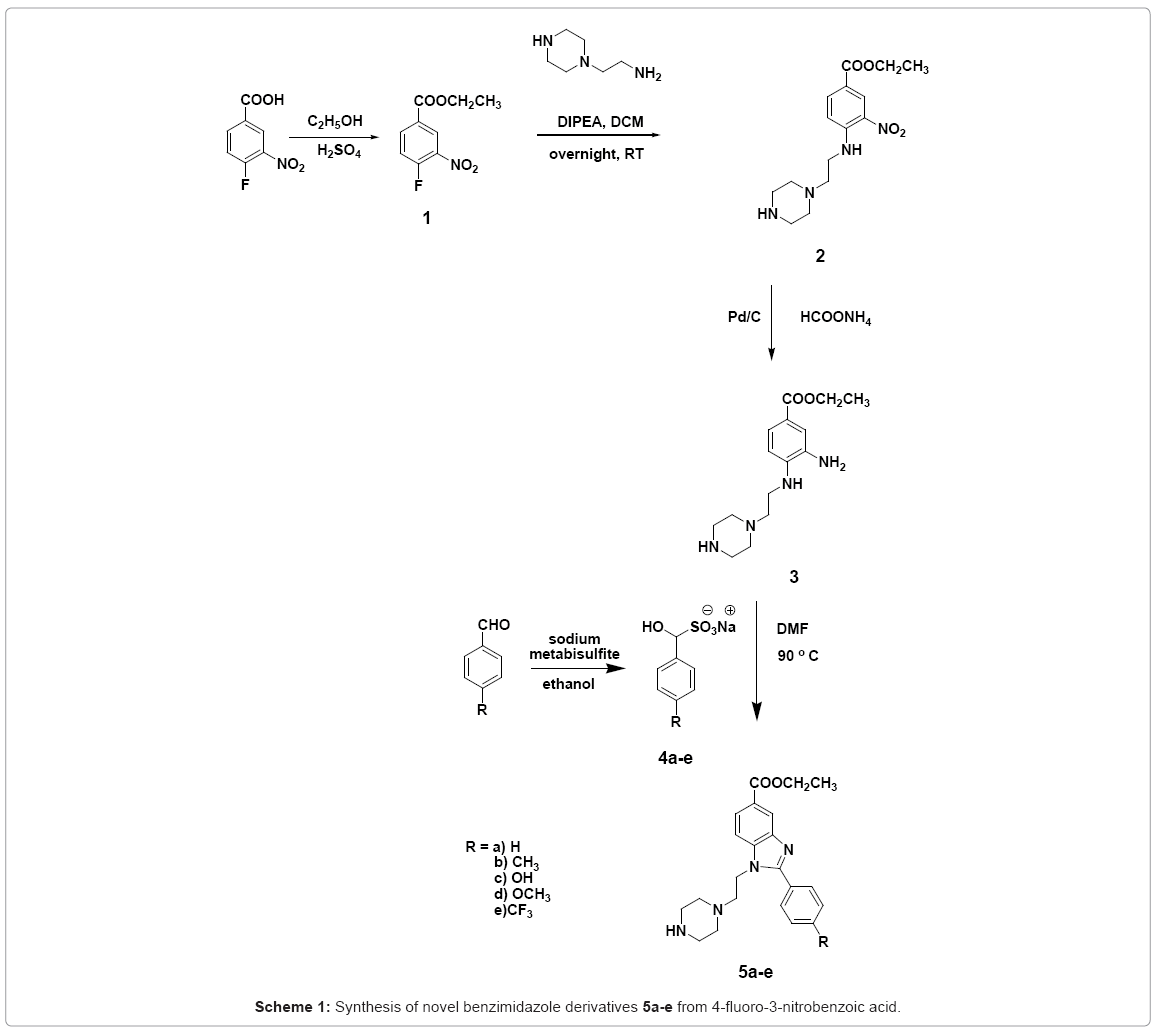
Scheme 1: Synthesis of novel benzimidazole derivatives 5a-e from 4-fluoro-3-nitrobenzoic acid.
Our synthetic study into polar benzimidazoles started with 4-fluoro-3-nitro benzoic acid which was esterified in the presence of catalytic sulfuric acid in ethanol by refluxing for 8 hours to afford the ethyl-4-fluoro-3-nitrobenzoate 1 in 75% yield. The ethylbenzoate 1. was then treated with N-(3-aminopropyl) piperazine and DIPEA in dry dichloromethane at room temperature to yield 2. However, our initial effort to synthesize ethyl 4-(2-(piperazin-1-yl) ethylamino)-3- nitrobenzoate 2 gave only 40% yield. The low yield of the product 2 led us to probe the reaction. Upon further analyses, we identified two side products from the reaction which were ethyl 4-(4-(2-aminoethyl) piperazin-1-yl)-3-nitrobenzoate, 2a and the diester product, 2b (Scheme 2).

Scheme 2: Reaction of 1 with N-(3-aminopropyl)piperazine.
This clearly showed that the reaction between a fluoro phenyl and amine happened at a very fast rate where no complete selectivity for primary amine over secondary amine was observed. This indirectly also implied that the probability of collision between the ethyl ester 1 and the reactant will determine the product(s) formed. Optimization to increase the yield of 2 was carried out and the results are presented in Table 1. No further efforts were made to determine the kinetics of the reaction.
| Entry | Conditions | 1 (mmol) | N-(3-aminopropyl)piperazine (mmol) | 2 (Yield,%) |
|---|---|---|---|---|
| 1 | Dropwise addition of N-(3-aminopropyl)piperazine to 1 | 1 | 1 | 40 |
| 2 | Dropwise addition of N-(3-aminopropyl)piperazine to 1 | 1 | 2 | 53 |
| 3 | Dropwise addition of N-(3-aminopropyl)piperazine to 1 | 1 | 4 | 71 |
| 4 | Reverse slow addition of 1 to N-(3-aminopropyl)piperazine | 1 | 1 | 56 |
| 5 | Reverse slow addition of 1 to N-(3-aminopropyl)piperazine | 1 | 4 | 89 |
Table 1: Results of the reactionof 1 andN-(3-aminopropyl)piperazine under various conditions.
The amino compound 2 was reduced to ethyl 4-(2-(piperazin-1-yl) ethylamino-3-aminobenzoate 3 using ammonium formate and 10% Pd/C for 1 hour to give 85% yield. We also tested the reduction reaction using sodium borohydride. However, comparatively, the yield obtained from NaBH4 was far lower (57%). This proved that palladium-catalysed transfer hydrogenation is an excellent method in reducing nitrobenzene to aminobenzene. This method is convenient, economical and uses a stable nonpyrophobic catalyst. The phenylenediamine 3 was then refluxed with various substituted bisulfite adduct of aromatic aldehydes 4a-e in DMF overnight to afford benzimidazole derivatives 5a-e in good to excellent yields. Among the literature reports available for the synthesis of benzimidazoles by the reaction of phenylenediamine with acid chloride [12], aldehyde [13] and acid [14], we found that access into benzimidazole derivatives via this metabisulfite route is efficient, environmental friendly and afforded good yield of the benzimidazoles.
The 1H NMR spectrum of benzimidazole 5a showed a singlet at δ 1.43 ppm due to the -CH3 from the ethyl group. The N-methylene protons on position-2 connected to the piperazine ring appeared as a double triplet at δ 2.78 and 3.50 ppm while the N-methylene protons from the piperazine also appeared as a double triplet at δ 2.24 and 3.11 ppm. The O-methylene protons (from the ester group) appeared as a quartet at δ 4.35 ppm. Similar 1H patterns were obtained for other substituted benzimidazoles derivatives 5b-e. The 13C NMR spectrum of 5a which resonated at δ 154.62 and 167.00 ppm are assigned to imine (C=N) and ester carbonyl carbon respectively.
The novel benzimidazole derivatives were subsequently assayed for their cholinesterase inhibition potency by Ellman’s method [15]. Results for their acetylcholinesterase (AChE) inhibition potentials are shown in Table 2. Rivastigmine was used as reference in the assays. We observed that electron withdrawing substituents at the R position in the phenyl ring are important for good activities as shown by 5e. The best inhibition was achieved by 5e with IC50 of 31.40 μM which was better than the standard drug rivastigmine.
| Compound | R | AChE inhibition (%)at 10 μM | AChE inhibitionIC50 (μM) |
|---|---|---|---|
| 5a | -H | 17.68 | - |
| 5b | -CH3 | 16.54 | - |
| 5c | -OH | 21.82 | - |
| 5d | -OCH3 | 24.77 | - |
| 5e | -CF3 | 40.09 | 31.04 |
| Rivastigmine | - | 32.28 | 45.15 |
Table 2: Inhibition of AChE by the synthesized compounds.
LogP/CLogP values [16] of the newly synthesized compounds 5a-e are shown in Table 3. Basically all of them fall in good range for prediction of drug activity and moderate toxicity. The tolerable toxicity of the compounds 5a-e was confirmed by the cytotoxicity test (IC50) in VERO cells at concentrationsup to 50 μM. After 72 hours of exposure, viability was assessed on the basis of cellular conversion of MTS into a formazan product using the Promega Cell Titer 96 Non-radioactive Cell proliferation method according to manufacturer’s protocol. All the compounds were found to be non-toxic up to 50 μM.
| Compounds | R1 | logP/CLogP |
|---|---|---|
| 5a | -H | 2.64/3.80 |
| 5b | -OH | 2.25/3.33 |
| 5c | -CH3 | 3.12/4.30 |
| 5d | -OCH3 | 2.51/3.81 |
| 5e | -CF3 | 3.56/4.69 |
Table 3: Lipophilicity logP/CLogP value of compounds 5a-e.
A series of novel polar benzimidazoles was successfully synthesized under mild reaction condition in good to excellent yield. Piperazinyl ethylbenzimidazole derivatives were derived from ethyl 4-(2-(piperazin- 1-yl) ethylamino-3-aminobenzoate with various substituted bisulfite adduct of benzaldehyde under reflux conditions. The synthesized novel polar benzimidazoles have potential biological applications such as therapeutics for Alzheimer’s disease in view of their good bioavailability. The bioactivity studies as well as quantitative structureactivity relationship of the newly synthesized polar benzimidazoles are on-going in our laboratory and would be published in the future.
The authors wish to express their gratitude and appreciation to Pharmacogenetics and Novel Therapeutics Research Cluster, Institute for Research in Molecular Medicine, Universiti Sains Malaysia, Penang for supporting this work. This work was funded through Research Grant No.RUC (1001/ PSK/8620012) and HiCoE research Grant No (311.CIPPM.4401005).