Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Editorial - (2012) Volume 1, Issue 2
Peroxidases (EC1.11.1.x) represent a large family of oxidoreductases that typically use hydrogen peroxide as an electron acceptor to catalyze the oxidation of substrate molecules. The vast majority of these enzymes contain heme as a cofactor [1], and are ubiquitously present in prokaryotes and eukaryotes. Peroxidases take center stage in a variety of biochemical processes, ranging from the biosynthesis of cell wall material to immunological host-defense responses [2,3]. Hemecontaining peroxidases were originally classified into two superfamilies: the plant peroxidases and animal peroxidases [4]. Remarkably, some members of the peroxidase superfamily have been studied for more than a century like, for example, Horseradish Peroxidase (HRP) [5], and in this respect, it was highly fascinating that the first member of a newly discovered peroxidase superfamily, the group of DyP-type peroxidases, was described in the late 90’s [6]. Here, we will discuss the biochemical and structural features of DyP-type peroxidases, as well as their promising biotechnological potential.
Dye-decolorizing (DyP-type) peroxidases were first discovered in fungi and are named after their ability to degrade a wide range of dyes [6]. Subsequently, additional members were found in the proteomes of other fungi, as well as in several bacteria [7]. This shows that these enzymes are widespread like other peroxidases. Interestingly, recent genome sequence analysis shows that these enzymes are prominent in bacteria, whereas only a small number is found in fungi and higher eukaryotes. Their occurrence in archaea is even more limited. The most comprehensive overview of the DyP-type peroxidase superfamily is offered by the Interpro database. According to this database, the DyP superfamily comprises currently almost 4000 members, of which 3707 are found in bacteria, 117 in eukaryotes and 11 in archea. The growing number of putative DyP-type peroxidases identified in the proteomes of bacteria, emphasizes our previous suggestion that this superfamily should be renamed into the superfamily of bacterial peroxidases [8]. Additionally, DyP-type peroxidases are, according to Peroxibase, further sub-classified into the phylogentically distinct classes: A, B, C and D. Many of the potential bacterial enzymes are putative cytoplasmic enzymes (class B and C), indicating that they are involved in intracellular metabolism. In contrast, enzymes belonging to class A contain a Tat-dependent signal sequence, which suggests that they function outside of the cytoplasm or extracellularly, as previously confirmed by us and others [8-10]. Class D contains primarily fungal variants. For some of these peroxidases, it has been shown that they are involved in dye decolorization [7]. Nevertheless, the physiological function of the majority of DyP-type peroxidases is at present unclear, although evidence is accumulating that some bacterial variants are involved in the degradation of lignin [8,11,12]. This suggests that these lignolytic enzymes can be regarded as the bacterial counterparts of the fungal lignin degrading peroxidases. DyP-type peroxidases are unrelated at the primary sequence level to peroxidases of the plant and animal superfamilies. They also lack the typical heme-binding motif of plant peroxidases, comprising one proximal histidine, one distal histidine and one crucial arginine (Figure 1) [2,5,7]. However, all DyP-type peroxidases contain the so-called GXXDG motif in their primary sequence, which is part of the heme-binding region. This motif is important for peroxidase activity because replacement of the conserved aspartate by an alanine or asparagine inactivates the enzyme, while heme-binding is not affected [8,13]. Based on these results, it was proposed that the conserved aspartate of the GXXDG motif is functionally similar to the distal histidine of plant peroxidases [1,2]. However, the catalytic role of this conserved aspartate was put into question by a recent study. It was shown that substitution of the aspartate of the GXXDG motif of E. coli EfeB/YcdB by an asparagine only, marginally affected the peroxidase activity of this enzyme [14].
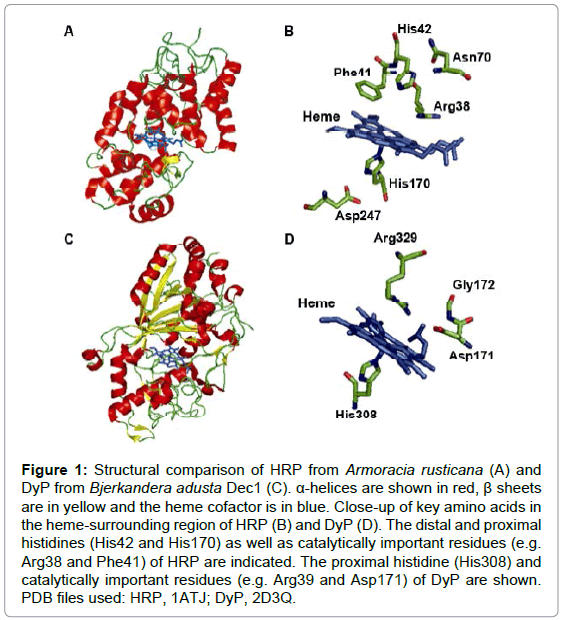
Figure 1: Structural comparison of HRP from Armoracia rusticana (A) and DyP from Bjerkandera adusta Dec1 (C). α-helices are shown in red, β sheetsare in yellow and the heme cofactor is in blue. Close-up of key amino acids in the heme-surrounding region of HRP (B) and DyP (D). The distal and proximal histidines (His42 and His170) as well as catalytically important residues (e.g. Arg38 and Phe41) of HRP are indicated. The proximal histidine (His308) andcatalytically important residues (e.g. Arg39 and Asp171) of DyP are shown. PDB files used: HRP, 1ATJ; DyP, 2D3Q.
A limited number of fungal and bacterial DyP-type peroxidases have been characterized in some detail, including elucidation of their crystal structures [13-16]. While DyP peroxidases from the different subclasses often exhibit a remarkable low sequence similarity, their overall structural topology is highly conserved. Structurally, DyP-type peroxidases comprise two domains that contain α-helices and anti-parallel β-sheets, unlike plant peroxidases that are primarily α-helical proteins (Figure 1). Both domains adopt a unique ferredoxin-like fold and form an active site crevice, with the heme cofactor sandwiched in between. The heme-binding motif contains a highly conserved histidine in the C-terminal domain of the enzyme (Figure 1), which seems to be an important heme ligand, and is therefore, functionally similar to the proximal histidine of plant peroxidases [13-16]. To test the role of the proximal histidine of DyP-type peroxidases as a heme ligand, we replaced this residue by an alanine in TfuDyP from Thermobifida fusca. This resulted in a loss of heme, which demonstrates that this residue is indeed an important heme ligand of DyP-type peroxidases [8]. In addition, fungal DyP peroxidases also contain a conserved histidine in the N terminal domain of the enzyme, which was previously assigned as heme ligand [17]. However, this residue does not contribute to heme binding, according to the available structures [13]. Clearly, more structural studies are required to unveil the molecular details by which DyP peroxidases catalyze oxidations.
The biochemical properties of only a few DyP-type peroxidases of fungal and bacterial origin have been analyzed so far [6,8,9,12,14-16,18-20]. These enzymes are typically 50-60 kDa, while several bacterial variants are somewhat smaller (about 40 kDa). All characterized DyP peroxidases contain non-covalently bound heme (proto heme IX) as cofactor, similar to peroxidases of the animal and non-animal subfamilies [8,9,13,15,21]. In addition, several oligomeric states have been reported, ranging from monomers to hexamers [6,8,9,14-16,18].
It has been well established that the catalytic mechanism of plant and animal peroxidases proceeds via formation of Compound I. It is, therefore, generally assumed that this is also the case for DyP-type peroxidases. Although the exact details about their catalytic cycle are still unclear, several recent studies point towards major differences between the catalytic mechanism of DyP peroxidases and other peroxidases. Based on four novel structures of a fungal DyP, it was proposed that the aspartate of the GXXDG motif swings into a proper position that is optimal for interaction with H2O2, thereby enabling compound I formation [22]. This crucial role of the conserved aspartate as a catalytic residue agrees well with the results of the mutagenesis studies on a fungal and a bacterial DyP, as discussed above. However, it is in contrast to, for example, plant peroxidases, where the distal histidine functions as an acid-base catalyst and compound I formation is assisted by an essential arginine (Figure 1) [5]. Furthermore, analysis of the peroxidative cycle of DypB from Rhodococcus jostii RHA1 established that its conserved aspartate is not required for peroxidase activity, because replacement of this residue by alanine had a marginal effect on the reactivity towards H2O2 and the formation of Compound I. Rather, a conserved arginine of DypB was found to be essential for peroxidase activity [23]. It therefore, appears that DyP-type peroxidases employ different residues as an acid-base catalyst during their catalytic cycle.
The most distinguishing feature of DyP peroxidases are their unparalleled catalytic properties. Firstly, these enzymes are active at low pH, which is most likely dictated by the aspartate of the GXXDG motif that functions as an acid-base catalyst at low pH, for at least a subset of DyP peroxidases [8,13]. Secondly, DyP peroxidases exhibit a unique substrate acceptance profile. These enzymes are able to degrade various dyes efficiently, and in particular anthraquinone dyes, which are poorly accepted by plant and animal peroxidases. Furthermore, DyP-type peroxidases display a poor activity towards azo dyes and small non-phenolic compounds, unlike plant and animal peroxidases [6,8,9,12,14-16,18,24-28]. Moreover, we have recently established that TfuDyP is able to oxidize aromatic sulfides enantioselectively, similar to plant peroxidases, thereby expanding their biocatalytic scope [5,8,29,30]. Intruigingly, DyP peroxidases appear to be multifunctional enzymes displaying not only oxidative activity, but also hydrolytic activity [31,32].
Plant peroxidases are attractive biocatalysts because of their broad substrate range, neutral pH optimum, and ability to catalyze reactions such as halogenations, epoxidations, hydroxylations and enantioselective oxidations, often accompanied with good yields [33]. However, the exploitation of these enzymes is hampered by their notoriously difficult heterologous expression and limited stability. With regards to the latter, it is interesting to note that DyP peroxidases appear remarkable robust, as shown by us and others [8,19,34]. Furthermore, our characterization of TfuDyP showed that this enzyme is expressed well heterologously in E. coli [8]. Combined, this underscores the biotechnological potential of DyP peroxidases, and shows that these enzymes, and in particular TfuDyP, are a promising alternative for known peroxidases. The potential of DyP peroxidases as useful biocatalysts for industrial applications is further emphasized by their ability to degrade a variety of synthetic dyes, indicating that these enzymes can be used for the bioremediation of dye-contaminated waste water. Moreover, it was reported that two fungal DyP-type peroxidases are able to degrade β-carotene [32]. The degradation of β-carotene is of interest for the food industry, enabling the enzymatic whitening of whey-containing foods and beverages. This specific application was patented recently, and the respective fungal DyP peroxidase is marketed under the name MaxiBright by DSM. The discovery of novel antimicrobial targets has become a pressing matter due to the vast increase of antibiotic-resistant, pathogenic bacteria [35]. With regard to this issue, it is important to emphasize that, as noted earlier, DyP peroxidases are remarkably abundant in the proteomes of bacteria, including many pathogenic bacteria, while these enzymes are absent in mammals. This indicates that DyP peroxidases could be promising, novel anti-microbial (pro) drug targets. This notion is supported by a recent study, which showed that a DyP peroxidase from Pseudomonas fluorescens GcM5-1A is toxic to cells of the Japanese black pine [36].
The group of DyP-type peroxidases comprises a newly identified superfamily of peroxidases, which are unrelated in sequence and structure to well-known peroxidases belonging to the plant or animal superfamilies. DyP peroxidases exhibit unique reaction features by displaying novel substrate specificities and reactivities. Additionally, DyP peroxidases can be remarkable robust and combined, this unveils their potential use as biocatalysts in a variety of biotechnological applications. However, these enzymes are only active under acidic conditions, which severely restrict their number of applications. It is therefore, desirable to alter their pH optimum by enzyme redesign to broaden their applicability. Conceivably, this could be achieved by constructing DyP variants, of which the active site more closely resembles that of plant peroxidases by replacing, for example, the conserved aspartate of the GXXDG motif with a histidine, because the pKa of the aspartic acid side chain is lower than the pKa of the histidine side chain. However, a recent study with such variants of DypB from R. jostii RHA1 showed that DyP peroxidases are unable to utilize the histidine efficiently as a proton acceptor, unlike plant peroxidases [23].
This clearly demonstrates that more rigorous engineering is required to obtain a pH-optimized DyP variant. Despite the promising biocatalytic potential of DyP peroxidases, much more work is needed to fully characterize the catalytic mechanism of DyP peroxidases, as well as the exact role of the catalytic residues, and in particular, the function of the conserved aspartate. Additional high resolution structures of DyP peroxidases from all the various subclasses are, therefore required, preferably in combination with different ligands. The limited number of DyP peroxidases characterized so far has established that these enzymes exhibit a vastly different substrate scope than plant and animal peroxidases, using however, a restricted set of diagnostic substrates. It is therefore, desirable that more and diverse substrates should be tested in order to fully understand their biocatalytic scope. Lastly, future studies should be aimed at investigating the potential of DyP peroxidases as novel microbial (pro) drug targets. In conclusion, it can be expected that the growing number of DyP-type peroxidases, biochemically and structurally characterized, will fully delineate their biotechnological potential. This will also provide new leads for the construction of improved variants suitable for biotechnological applications.