Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2013) Volume 6, Issue 8
Multifaceted data scattered among numerous repositories became a reverse of the medal in the postgenome era. Withstanding information pieces are aggressively crawling away, we proposed a Web-content management system–genoCMS. The system designed to “domesticate” data locally by sampling genes and re-annotating them in a user defined manner. Heat map visualization style was implemented in genoCMS to make it easier for user to inspect data coverage across a collection of genes. Using genoCMS, we have revisited the recently published data on large-scale transcriptome and proteome mining. Demonstration scenarios were implemented to illustrate the sortand- select workflow for distilling genes with desirable profiles of the features.
Keywords: Knowledge base; Heat map; Omics science; Integrative bioinformatics; Human Proteome Project
The gap between “omics” and biochemist’s viewpoint is vividly depicted, as biochemists rarely read the articles written by highthroughput researchers. There is a need for the translation of accumulated multi-omics datapoints into the transparent message for practical biochemists. There should be a new generation of researchers that would interface “The Omics Factory” and “The Lab” and close the chasm between the hypothesis-free discovery and hypothesis-driven dissection of biological systems [1]. This foresight envisions a novel type of scientific interface software, assisting the generation of the “ready-to-go” testable hypotheses available for immediate uptake by scientific community for subsequent experimental testing.
Conversion of data acquisition mode into the hypothesis-driven data processing is a mainstream of the chromosome- and gene-centric approaches promoted by Chromosome-centric Human Proteome Project (C-HPP) [2,3]. Seeking for an effective way to represent and share the data, the heat map view was elaborated by C-HPP teams as a simple tool, easily supported by a spreadsheet, like Excel [3,4]. The heat map is generated by listing the gene names in rows and their features in columns (or vice versa). Each cell is color-coded in accordance with its numerical value. Color coding is performed according to the indicative scheme to enable visual perception of the statistical properties of the underlying data.
The C-HPP provides an approach for data “domestication”, a rationale behind the development of the reported system. We observed, that although the powerful data integration tools are available (Taverna [5], Galaxy [6], BioUML [7], Genome Browser [8]) together with specialized human protein resource NeXtProt, the Chr-centric research teams of HPP proposed their own Web-solutions. The Proteome Browser (TPB [9]) piloted in 2012, enables the organization of multiple tracks into the hierarchical structure. The Chromosome-assembled Proteome Browser (CAPER [10]) supports both a traffic light and heat map method of data depiction. The Genome-Wide Protein Database is a specific resource to observe the human placenta protein profile of whole human genome (GenomewidePDB [11]).
Why the same wheel was reinvented at least three times during just one year since C-HPP beginning, despite flexible tools for workflow management and information browsing were readily available on Internet? We proposed that development of the Web-applications is a contemporary form of natural understanding of the complicated postgenome data. The data itself as well as Web-based sources reached such degree of sophistication, that deep and precise vision can be achieved by re-constructing of the data array (at least partially). To understand the whole, the bioinformatician has to reproduce some part in own hands.
The similarity of the approaches taken by different C-HPP teams for data domestication is really amazing. The current versions of proteome browsers on the Web integrate information from generally the same public Web-resources, adopted by C-HPP. Heat mapping shows the UniProt evidence level, in accordance with the expert assigned status: “protein”, “transcript” or “predicted”. The UniProt data is also recruited to delineate features such as the number of nonsynonymous polymorphisms (nsSNP) and number of alternative spliced transcripts (AST). Secondly, the mass-spectrometry (MS) information is incorporated into the heat map to reflect either the number of observations or the quality of protein identifications. This type of information is typically retrieved from three complementary resources of tandem MS (MS/MS): PRIDE [12], PeptideAtlas [13] and GPMdb [14].
To tackle the problem of data domestication we developed the Web-based content management system (genoCMS) for rapid genecentric consolidation of the data. We show how genoCMS can be used to accommodate the tracks selected by C-HPP protein browsers [9-11]. Also genoCMS is helpful to compare public data against fresh unpublished results, supporting the multi-laboratory research [15]. The use of our system has been demonstrated on chromosome 18, the Russian contribution to the Chromosome-centric HPP [16].
Initial setup
GenoCMS was installed and, to customize the system for this study, the initial set of 277 genes from chromosome 18 was loaded. The list of entries for the target chromosome was obtained by entering the relevant query into the UniProt search interface (UniProtKB/Swiss- Prot, release 2012_07). The retrieved XML file was imported into genoCMS using the following fields: UniProt accession code, gene symbol with alternate names, and protein full name.
The setup procedure included the creation of data tracks by manual input of full name, short label and description. In total, 17 tracks were created specifically for this study (see Table 1), but additional tracks were also generated to meet the specific tasks of the on-going HPP. For each track the data source was specified as either Web-service or biocuration. Web-service automatically computes and returns, on request, the numeric value of the feature. The biocurated track is imported as a comma-separated spreadsheet and can be manually corrected by an expert.
| No | Track Short label | Source (version) | Type | Value | Chromosome 18 coverage, % | Update mode1 |
|---|---|---|---|---|---|---|
| 1 | HepG2_iBAQ | Geiger et al. [17], MaxQB (3.3.6.1) [18,19] | Continuous, quantitative | iBAQ index of protein copy number per HepG2 cell | 30.3 | BC |
| 2 | Liver_RNASeq | RNAseqAtlas (nov.2012) [20] | Continuous, quantitative | Liver expression marked RPKM (reads per kilobase per million) numbers for the liver | 71.1 | WS |
| 3 | Cancer | GeneCards (3.09 18 Nov 2012) [21] | Continuous, quantitative | Score (proportional to number of papers supporting gene association with cancer) | 18.1 | BC |
| 4 | Dr | OMIM database (November 30, 2012) [22] and by text-mining of articles [23]. | Continuous, quantitative | Number of diseases associated with protein | 10.8 | BC |
| 5 | Evidence | NeXtProt (rel.2012-10-07) [24] | Discrete, qualitative | Evidence: protein level, transcript level, predicted/homology and uncertain | 100 | WS |
| 6 | PlasmaAbund | PeptideAtals (2012_07) [13], Farrah et al. [25] | Continuous, quantitative | Number of protein spectra | 7.6 | BC |
| 7 | Plasma3020 | Omenn et al.[26] | Discrete, qualitative | Abundance in plasma: protein observed | 11.9 | BC |
| 8 | BioMRM | Hüttenhain et al. [27] | Discrete, qualitative | SRM assay developed, protein detected in plasma | 0.7 | BC |
| 9 | MSDB | PRIDE (v2.8.17) [12], PeptideAtlas (2012_07) [13], GPMdb (rel.2012-09-01) [14] | Continuous, quantitative | Normalized number of observations in MS experiments | 99.6 | WS |
| 10 | PA_supp | ProteinAtlas (20120_07) [28] | Discrete, qualitative | Indicated the degree of supportive information for the protein-specific antibodies: immunohistochemistry, western blotting, immunofluorescence, protein array | 71.8 | WS |
| 11 | PPI_STRING | STRING (9.05) [29] | Continuous, quantitative | Number of interacting partner for a gene product | 87.7 | WS |
| 12 | PPI_IntACT | IntAct (v.4.1.0 rel 168) [30] | Continuous, quantitative | Number of interacting partner for a gene product | 63.9 | WS |
| 13 | PPI_BioGRID | BioGRID (3.2.102) [31] | Continuous, quantitative | Number of interacting partner for a gene product | 69 | WS |
| 14 | PPI_HPRD | HPRD (rel.9) [32] | Continuous, quantitative | Number of interacting partner for a gene product | 50.2 | WS |
| 15 | PPI_MINT | MINT [33] | Continuous, quantitative | Number of interacting partner for a gene product | 29.6 | WS |
| 16 | VC-IP | GPMDB (rel.2013-08-01) [14] data processed algorithm proposed by Zhang et al. [34] | Continuous, quantitative | Number of interacting partner for a gene product obtained using method virtual co-precipitation | 40 | BC |
| 17 | PPI_Cont | CRAPome [35] | Continuous, quantitative | Data of common contaminants in AP-MS experiments | 41.9 | BC |
1BC – biocuration. WS – Web service.
Table 1: Description of the tracks.
To complete the setup procedure the tracks were organized into three categories. The first data-mining category encompassed the tracks with values automatically retrieved from public repositories, including NeXtProt, ProteinAtlas [28], GPMdb [14]. The second experimental category was for the data arrays extracted from the supplementary materials of papers. The third predicted category consolidated the tracks with values created using computational algorithms. Generally, genoCMS supports an unlimited number of categories for the tracks.
Sources of information for the tracks
The tracks descriptions and corresponding sources - databases and papers - are listed in Table 1. The data for genoCMS was either uploaded automatically from public sources by using web service (WS in Table 1) or loaded manually from supplementary materials of recent articles in biocuration mode (BC in Table 1). We employed tracks 1-10 for use case #1 and use case #2 was illustrated with tracks 11-17.
The data from three MS/MS repositories named PRIDE, PeptideAtlas and GPMdb were combined into single MSDB track according the formula:
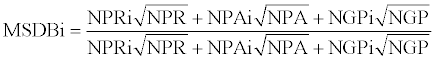
where MSDBi is a track value for i-th protein, NPR, NPA, and NGP-collection of number of protein identifications in PRIDE, PeptideAtlas and GPMdb respectively, - the arithmetic means denoted by a bar.
- the arithmetic means denoted by a bar.
Using the computational algorithm proposed by Zhang et al. [34] and the GPMdb as a source of MS/MS data the number of interactors was computed. The results were uploaded to genoCMS as “Virtual Coprecipitation PPI” track, briefly VC-IP.
Supplementary data from published papers were processed to map the identifiers to the UniProt accession codes. All files were prepared in the comma-delimited format and imported to genoCMS. Records for genes that were not listed as the target set of chromosome 18 genes were discarded during the import process.
Color schemes
The initial color schemes were specified for each track during the genoCMS setup. The color schemes were further corrected after the track values for each gene were uploaded into the system. The histograms of the values of features were used to elaborate the color schemes. The colors were dependent on the distribution of the features’ values of a track. Five ranges were generally used, each absorbing approximately 20% of the total amount of the sampled values. The values of qualitative tracks, e.g. NeXtProtprotein evidence, were assigned to the integers, each of which was further associated with a distinct color. The status of “no data available” remained in black.
Workflow
The system supports a sort-and-select workflow that consists of iteratively repeated operations. First, the tracks are selected to be displayed on the heat map. The selection of a limited number of tracks is based on the intention of the user to observe the general overview of the whole dataset on one page. Secondly, the heat map is sorted by the track values. The portion of the sorted genes is selected and moved to a separate gene set. At the next iteration the new tracks are chosen specifically for the created gene set.
Implementation details
GenoCMS is structured as a three-tiered application consisting of data, application logic, and presentation. The application logic tier is implemented in Java(v1.7) and Groovy using the Grails(v2.0.4) web framework extended the RESTful BioCurations Developer’s library. The data tier is a relational database managed by PostgreSQL. The presentation tier is comprised of dynamically generated html pages and JavaScripts. The system operates under the Apache Tomcat web server. Web services that are compliant with genoCMS are developed in Python and deployed with Google App Engine.
The customized chromosome 18 version of genoCMS is accessible at www.kb18.org. Registration is required for saving sets and creating proprietary tracks. The genoCMS package is freely available on request as a compressed virtual machine.
Up to 20 thousands of objects (genes) are technically supported by genoCMS, making it possible to apply the system to different human chromosomes as well as to the genome-wide set of protein coding genes.
We have developed genoCMS - a web-based content management system to support the collaborative discovery over the collection of genes by using their color-coded annotations (Figure 1). The heat map view enables observation of the color of tracks across the genes and allows tracks to be toggled on and off. Genes can be sorted according to the value that has been loaded for the tracks. Genes with distinguishable profiles of features can be selected by the user. Selected genes can be copied to a separate set for the next round of work.

Figure 1: Heat map view of a collection of genes: (a) – colored-coded tracks; (b) – checkbox for selecting the genes; (c) – add-to-set \ delete-from-set buttons; (d) – export to spreadsheet and (e) – add \ remove tracks from the map.
Uploading the data into genoCMS
Gene-centric CMS was populated with the data shown in Table 1. A total of 17 tracks were loaded from different sources. The core tracks were taken from the public databases, recommended by C-HPP [2-4,36]. Other tracks were assigned the values using information from a number of the seminal papers in modern proteomics (see Table 1). Tracks were unequal in terms of coverage of chromosome 18 genes, with the least coverage observed for the proteins detected in biofluids by either MRM (BioMRM) or MS/MS (PlasmaAbund).
A relatively high coverage of 71.7%, 71.8% and 87.7% of chromosome 18 genes was observed for RNA-seq Atlas, PA_supp and PPI_STRING tracks, respectively.
The average of the MS data from PRIDE, PeptideAtlas and GPMdb was composed into the single track, named MSDB. Almost complete coverage of the chromosome was achieved, as shown in Table 1. However, the quality of many of the rare observations was questionable.
A number of tracks called “discrete” in Table 1 enabled the qualitative descriptors to be displayed in traffic light coloring. For example, using ProteinAtlas it is possible to estimate the quality of the antibody, using immunohistochemistry, western-blotting, immunofluorescence and protein array data [37]. These estimations were colored red when taken from one source of supportive data, and yellow or green for two or more simultaneous sources of supportive data, respectively. Similarly, the evidence track taken from NeXtProt, as protein level, transcript level, predicted/homology and uncertain, was coded in green, yellow, orange or red, respectively.
Traffic lights are also of use to map the published data in the cases when the categories are not clear, as shown for the BioMRM track (Table 1). The values of this track originate from the supplementary materials on the development and detection of multiple reaction monitoring assays for more than 1000 cancer-associated proteins [27]. We depicted the availability of the MRM assay in yellow, when assay was elaborated using the MS/MS analysis of the mixtures of crude synthetic peptides. The green color was used to indicate if proteins were measured in human plasma by the multiple reaction monitoring.
In contrast to the discrete traffic light coloring, a continuous color scheme was applied to the naturally numerical source of the data. In Table 1 the continuous quantitative tracks are exemplified as iBAQ proxy of protein abundance in HepG2 cells. Other quantitative tracks represent mRNA expression level in liver and frequency of protein occurrence in the MS/MS datasets of protein identification (MSDB).
Assigning color scheme for the tracks
Due to uneven nature of the post-genome data it is challenging to recommend single approach to assign colors for the values in the track. To show one of the possible approaches the data on protein abundance has been taken from the large scale MS/MS experiment [38]. The idea was to illuminate, if there exists the MS/MS quantified proteins, which levels change by an order of magnitude. The iBAQ index for cancerous tissue was normalized to the corresponding index in the normal biopsies. The distribution of the log-ratio is shown in Figure 2. From the distribution a rare cases are observed, when the protein level is changed 10 times. So, the yellow color is assigned to indicate the importance for the events with the log-ratio above 1 (10-fold increase) or -1 (10-fold decrease). Most of the values occupy the range of colors reduced to grey as this color merges to the background color of the heat map. The inset in Figure 2 illustrates how genoCMS enables to adjust the color scheme for introducing the subjective vision of the underlying data.

Figure 2: Assignment of the user-defined color scheme using the distribution histogram. Ratio.C.N. -- Ratio of label-free protein abundance estimation from shotgun MS/MS data for normal colon tissue (N) and adenocarcinoma (C) [38].
Use case #1-selecting the salient genes
When tracks were loaded with the appropriate color coding, we implemented the use case restricted to the target set of chromosome 18 genes. The demonstration task was to sort out a number of salient proteins that were potentially relevant as cancer biomarkers.
As the first step, the set of target genes was visualized on the heat map. Two tracks were initially taken one for protein abundance (iBAQ index) in the HepG2 cell line and the second for transcript abundance (RPKM) in liver tissue. The map was sorted according to the HepG2_ iBAQ track. The top 14 genes with iBAQ indices from 6.70 to 7.92 were observed on the sorted heat map. Those with low RPKM values were selected. These were visualized as genes neighboring those in green for protein abundance and genes in yellow or red for transcript abundance.
The second step was to sort the heat map in descending order of transcript abundance, with the 14 low-abundant proteins at the top. This fraction was also sorted to collect the nine cases with a discrepancy in the colors of the neighboring panels, e.g. yellow or red for protein abundance and green for transcript abundance. This resulted in a list of 18 genes/proteins that are differentially expressed in HepG2 and liver tissue.
The comparison of differential expression was made at the protein level for HepG2 cells and at the transcript level for the liver tissue. In the rigorous sense such a comparison is inadequate, but taking in consideration the sufficient degree of correlation between RNA seq and MS/MS data [39], it could be accepted as a preliminary approach to chromosome data analysis.
The last step was capturing the distilled genes into a separate and “switch on” additional tracks to expand their characteristics (Figure 3). The set was sorted by the MSDB track to rank the genes according to the accessibility of their protein products by means of MS. The relevance of the differentially expressed genes to cancer is shown for five genes: TTR, MAPRE2, TXHL1, EPB41L3 and MIB1 (Figure 3). For example, the expression of transthyretin (TTR) in liver is higher than in the cancer cell line, whereas for the TXHL1 gene the opposite situation is observed.

Figure 3: Final set of the salient genes, potentially relevant as cancer biomarkers, that were distilled via sort-and-select workflow. Presented genes are characterized by the differences in HepG2_iBAQ and Liver_RNASeq tracks.
With an exception of two genes, the remainder shown in Figure 3 are amenable to detection by antibodies, as indicated by ProteinAtlas data (PA-supp track). The green color of this track indicates that the quality of the antibodies was confirmed by several supportive sources.
The data on the biofluid availability of the genes is presented in Figure 3, by three tracks: PlasmaAbund, Plasma3020 and MRM. Only TTR protein was measured by MRM in plasma, as reported by Hüttenhain et al. [27]. The rarity of the measurement tells us that, due to the limitation of sensitivity of MS, most of the cancer-related proteins remain undetectable in a single set of experiments used for targeted analysis [40]. Combination of the multiple experimental sets from Farrah et al. [25] and Omenn et al. [26] made additional proteins in human plasma available for heat map analysis. These proteins included DSC2, MYO5B and USP14.
Use Case #2-cross-database comparison
The task was to evaluate the large data repositories as a source of protein-protein interactions. By using sort-and-select a portion of genes was elaborated and assembled into the separate set, shown in Figure 4. Genes having values for VC-IP tracks were selected. The protein coding genes missing in at least one of the interactome databases were excluded from the set. This generation resulted in the set of 32 genes. The genes occurring in more than 10% affinity purificationmass spectrometry datasets were discarded using the track PPI_Cont. The final set contained 19 protein coding genes (Figure 4). Finally 11 proteins were distilled concordant in the number of PPI across all the databases and characterized by low (<3.5%) contaminant probability (selected in Figure 4b).

Figure 4: Comparing number of partners for Chr 18 genes available in PPI databases using the matrix (A) and table (B) views, supported by genoCMS.
For SMAD2 and SMAD4 genes all cells of the heat map are green, indicating that each database contains relatively high number of interactors, e.g. 129 for SMAD2 in the case of BioGRID (see tooltip in Figure 4a). The absolute number of interactors can vary across databases, as shown using the genoCMS tabular view in Figure 4b. STRING contains maximal number of interacting partners for almost all of the analyzed genes, with exception of ubiquitin-protein ligase NEDD4L, which has 113 partners according to BioGRID versus only 47 partners in STRING. Analysis of the table view across databases enabled to indicate SMAD2, SMAD4, PSTPIP2, NDUFV2, GATA6, MBD1, SMAD4, PIK3C3, LAMA3, MALT1 and RALBP1 as essential proteins on Chr 18. Such essential proteins can be recommended as baits for the affinity-purification MS.
We presented a genoCMS, the Web-based Content Management System, created to meet the challenge of biomedical data domestication. This tool is used to generate a new datasets from the data of many sources, which is then used to select relevant genes. The applications of genoCMS are compliant with the motto of the Chromosome- Centric Human Proteome Project. GenoCMS enables to get focused on a limited number of genes and to compile the relevant information across the data repositories.
Two use cases for genoCMS were presented. Both served to develop a limited list of the genes/proteins, which are important to formulate the hypotheses for experimental testing. The application of the system was illustrated by processing the data arrays from several recent papers on deep proteome mining, targeted plasma profiling and RNA seq experiments. A set of salient proteins was distilled out of the current compendium of chromosome 18 genes using the sortand- select workflow. Working within the restricted domain of a single chromosome, the system delivered an approach to track sparse and hardly comparable data.
Gene-centric CMS provides a mechanism to introduce new tracks, which is useful whenever datasets become published on Web. The tracks registered in genoCMS are automatically subjected to periodical updates.
In contrast to existing proteome browsers [9-11], genoCMS was destined to support the collaborative work by creation of sets assigning the tracks. As web-services appear, these can be registered within the system as new tracks. Experimental arrays can be appended as dedicated tracks via the biocuration mode. Once registered, the track becomes available for all of the genoCMS users and can be combined with other tracks to expand the heat map. Registered genoCMS user can establish the set of genes, decorate it with previously existing and original tracks. The decorated set is shared among the participants of coordinated project.
The work has been supported through a State Contract with the Ministry of Education and Science #2002. Authors are grateful to Ancha Baranova (Research Center for Medical Genetics, Russian Academy of Medical Sciences, Moscow, Russia; School of Systems Biology, College of Science, George Mason University, Fairfax, VA, USA) for inspiring the idea of the manuscript.