Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2017) Volume 4, Issue 2
Chronic kidney disease (CKD) is becoming an extensive public health problem worldwide. The current anxiety of disease might be due to the change of the underlying pathogenicity. The aim of our study was to provide a detailed analysis of microarray gene expression data of CKD in correlation with diabetes and identification of biomarker genes. Here, Affymetrix expression arrays were used to identify differentially expressed genes in 22 and 69 samples of CKD and diabetes respectively. It further outlines few of the principal biological alterations observed in the CKD state and depicts specific procedures for conducting quality assessment of Affymetrix Gene chip using GEO datasets (GSE70528, GSE11045) and also illustrates quality control packages to remark the visualization for detailed analysis. We identified 912 differentially expressed genes in CKD and 629 in diabetes. From extensive comparison of CKD with diabetes, we found 80 common genes, of which 29 were found up regulated and 51 down. Further, analysis with NCG of these 80 genes, 10 common genes were found involved in various types of cancer. Thus, the results emphasize the importance of these 10 common differentially expressed genes in considering them as biomarkers for three conditions diabetes, CKD and cancer. Our studies have cataloged differentially expressed genes that may play important role in the pathogenesis of CKD and could serve as biomarkers.
<Keywords: CKD; GEO datasets; Diabetes; Kidney; DEGs
Chronic kidney disease (CKD) is chronic, non-communicable, age related renal function decline disease that affects people worldwide [1]. The prolonged CKD is a major risk factor of cardiovascular diseases and death [2]. The disease prevalence increased with age and gender specific affecting more females than males. The healthcare burden is highest in early stages due to increased prevalence, affecting around 35% of those over 70 years [3]. The unhealthy lifestyle, diabetes, hypertension, BMI, obesity and smoking are some major factors that accelerate CKD. Since, high blood pressure and hypertension are the major causes of kidney failure as these provide stress on blood vessels throughout the body, including nephrons [4]. Further, increasing cases of diabetes complicates the CKD disease thus making it healthcare priority in both developed and developing countries. CKD symptoms include abnormalities in kidney structure and function. It can be manifested with or without decrease in GFR that is considered to be as the markers of kidney damage [5]. Late appearance of symptoms makes it hazardous, often causing irreparable damages to the kidney requiring expensive treatments and renal transplant [6]. According to Global Burden of Disease report 2015 of ISN (International Society of Nephrology’s) [7], it is reported that the CKD was ranked 18th, which is second only to that for HIV and AIDS [8]. The overall prevalence of CKD is 13% to 25% as calculated for the world population [9].
Diabetes accounts for approximately 2% death of patients starting treatment for kidney failure each year, including dialysis and the renal transplantation for both type 1 and 2 [10]. Nearly 44% of dialysis patients have Type 2 diabetes, making it the fastest growing risk factor for CKD. Type 2 diabetes is the number one cause of kidney failure, responsible for more than one of every three new cases [11]. CKD emerged as a global burden requiring attention in understanding the disease dynamics rendering prevention and early detection as a health care priority in both developed and developing countries. Novel and affordable treatment strategies, identification of prognostic biomarkers for progression and treatment response are urgently required for this endemic disorder [12].
High throughput gene expression profiling has emerged as a dynamic research tool that uncovers transcriptional patterns in numerous biological systems to acquire insight into disease dynamics [13]. The development of high-throughput microarray technology, permitting concurrent measurement of changes in expression of multiple genes within the human genome, equips the opportunity for novel insight into disease processes and molecular pathways of biological dysfunction [14]. A vital, yet challenging arena is quality control and visualization of microarray gene expression data. One of the most prominent platforms for microarray analysis is Bioconductor, an open source and open development software project for the analysis and discernment of genomic data [15].
The aim of our study was to provide an unbiased comprehensive catalog of change in gene expression for the human CKD samples. We tried analyzing and visualizing the differentially expressed genes in CKD together with the effect and correlation to the Diabetes type 2. Here, we presented a gene-expression analysis of CKD (22 samples each) from healthy persons and patients. The heuristic comparison of three conditions CKD, Diabetes and Cancer has revealed the occurrence of 10 prominent genes. These genes are required for the basic functioning of the cell like in the rearrangement of the cytoskeleton, activation of immunity and development of tissues and kidney. So, the Up regulated genes (4 in number) directly confer the increase in the conditions like leukemia (increase in WBC) and alter the immunity by hampering the T-cell maturation. Similarly Down regulated genes (6 out of 10) disturbs the development of kidney i.e., the formation of new tissues and also disturbs the GFR (effect of the Osmotic pressure) that directly effects normal functioning of the kidney. Thus, this study emphasize the importance of these 10 genes (both Up and Down regulated) as biomarkers for three conditions CKD, diabetes and cancer. We also expect that this work will benefit the researchers working in in-vitro to collect evidences against CKD.
Microarray data
The framework for microarray analysis is shown in the Figure 1. The gene expression data bearing GEOID: GSE70528, GSE11045 and GSE30122 (contain 69 cel files with diabetes) were retrieved from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) [16]. The GSE70528 RNA microarray analysis was performed on 19 patients without diabetes. These patients were categorized into three groups: HT (8 hypertension patients), CKD (7 patient’s not on dialysis) and HD group (4 patients Haemodialysis). We extracted three normal CEL files from GSE11045, thus, total 22 CEL files. In addition, we also retrieved gene expression data for capturing the scenario in diabetic patients.

Figure 1: Flowchart of microarray data analysis.
Microarray analysis
The data fetched above was then subjected to R packages given in Table 1. According to the corresponding correlation between the probe and gene from the GPL570 platform HGU 133 Plus 2 (Affymetrix Human Genome U 133 plus 2.0 Array, Affymetrix, Santa Clara, CA, USA) [17]. The probe numbers of the expression profile were converted into the corresponding gene symbols. One gene corresponds with multiple probes, thus each gene has more than one expression value. Therefore, the average value was calculated and selected as the only representative value. A total of 54675 genes were involved in preprocessed data. Background correction and quartile data normalization were performed by the robust multi-array (RMA) average in R affy package (Affymetrix). In pre-processing, convolution background was corrected, missing values were estimated, expression values were log2 transformed and then normalized. Both data cel files pre-process under the MAS5 method [18]. The expression value after summarization is further use for DEG’s.
| S. No. | Package | Description |
|---|---|---|
| 1 | Affy | Exploratory oligonucleotide array analysis |
| 2 | AffyExpress | Quality assessment and to identify differentially expressed genes in the Affymetrix affymetrix chip |
| 3 | AffyQCReport | Quality of a set of affymetrix arrays |
| 4 | AffyRNADegradation | Assessment and correction of RNA degradation effects in Affymetrix 3' expression arrays |
| 5 | ArrayTools | Quality assessment and to detect differentially expressed genes for the Affymetrix GeneChips |
| 6 | DEGseq | To identify differential expressed genes from rna-seq data. |
| 7 | DEGreport | Report of deg analysis |
| 8 | Simpleaffy | High level analysis of Affymetrix data |
| 9 | AffyPLM | Quality assessment tools for affymetrix chip |
| 10 | Affy comp | Compare expression measures for Affymetrix Oligonucleotide Arrays |
| 11 | Derfinderplot | To find the plot for derfind. |
| 12 | Affyio | Parsing Affymetrix data files |
| 13 | ArrayQualityMetrics | Microarray quality metrics reports |
| 14 | AffyPLM | Quality assessment tools for affymetrix chip |
Table 1: Analyzing and Visualizing of CKD Data Using Quality Control Packages of Bio-Conductor.
Visualization plots
For visualizing of chips affyqcreport package was downloaded from Bioconductor [19]. Starting from library affy package and readaffy function read the 22 samples from directories celfiles names save as in the same directories having all plot described below. There are number of plots built in quality control packages for visualization and analysis of chips. These plots were also used for comparing chips before and after normalization [20]. Here we describe some plots and their analysis.
Box plot: Boxplot also called as box-and-whisker plot, is statistical plot that graphically plots numerical values for comparison of chips. Five values were plotted in box plot that are lower min value, lower quartile, mean, upper quartile, max value of chip files in parallel for comparison of chip intensities.
Intensity plot: Intensity plot is similar to Boxplot but it gives more detailed view. Intensity plot x-axis represents probe density and y-axis probe intensity.
MA plot: MA plot is used to determine if there any biasness in intensities of red and green dye and other systemic errors or instrumental errors in experiment.
RNA Degradation Plot: RNA degradation plot is used to assess the quality of RNA molecule used as probe in chips. This plot is used for quality analysis of probes spotted on affymetrix chip.
NUSE plot: Normalized Unscaled Standard Errors (NUSE) is used for standard error for each probe on all chips. All probes are normalized to scale one across all arrays.
QC stats plot: QC plot is recommended by affymetrix. Any array that is shown in red is indication of error and in blue indicates the array within limits of scale factors e within 3-fold.
Median Absolute Deviation (MAD): The low or high median of the absolute deviations was computed from the median and adjusts by a factor for asymptotically normal consistency.
RLE plot Relative Log Expression: RLE plot is a plot of RLE values that are calculated by assuming that all probe set across arrays against median expression value is constant.
This study provides information on the structure of correlation based tuning between genes in multiple microarray data sets. Our main goal was to establish the set of Fundamental genes by comparing analyses across data sets that is relevant in understanding gene function. Thus it was found that many genes showed patterns of correlated expression that are reproducible across data sets, and there is a clear relationship between confirmation of correlated expression and related gene function. From GSE70528 data, total 19 cel files are selected out of which 8 hypertension data, 7 CKD data and 4 hemodialysis data chip used in this experiment is HGU 133 Plus 2. On the other GSE11045 from GEODATASETS3 normal kidney cel files downloaded and taken into cel files folder. Gene expression file is created using affy package of bioconductor and visualization packages are used for quality control analysis and summarizing the output generated by these packages [21].
Visualization plot
Gene expression file created using affypackage.
>library (affy)
>cirbs=Read Affy (changedir ≤ CELFILES)
>eset.mas5=mas5 (cirbs)
>exprSet=exprs (eset.mas5)
>exprSet=log (exprSet,2)
>write.table (exprSet,file=”myab”, quote=F, sep=”\t”)
We used rmaan another method which create expression file for visualization plots
>eset=rma (cirbs)
>write.exprs (eset,file=”cirbs.txt”)
Using these above commands in R, the file containing the expression data can be created and will be used for further analysis using statistics. The object ‘exprs’ created in R for expression file using the simple command. This object exprs can be used directly in R to visualize expression file and analyze the intensity [22]. Four Affymetrixspecific metrics were evaluated if the input object is an AffyBatch. The RNA degradation plot from the affy package, the relative log expression (RLE) boxplots and the normalized unscaled standard error (NUSE) boxplots from the affyPLM package and the QC stat plot from the simple affy package are represented [23]. For visualizing data given by affyQCReport an R package the following set of command are required.
>Library (QC Report)
>QCReport(data, file=”cirbs.pdf”)
Boxplot: Each box shows the distribution of expression values of one array. The shape of the box plot for all the arrays looked similar, thus we concluded that there were less systematic biases in the data. The box plot of raw intensities of the data across the 22 chips is depicted in see Figure 2A. The raw intensities differed between HT 1 and HT 2, CKD 6, CKD 7 and HD 4.

Figure 2: A. Boxplots for arrays GSM1808377.HT-01, GSM1808378.HT-02, GSM1808379.HT-03, GSM1808380.HT-04, GSM1808381.HT-05, GSM1808382. HT-06, GSM1808383.HT-07, SM1808384.HT-08, GSM1808385.CKD-01, GSM1808386.CKD-02, GSM1808387.CKD-03, GSM1808388.CKD-04, GSM1808389. CKD-05, GSM1808390.CKD-06, GSM1808391.CKD07, GSM1808392.HD-01, GSM1808393.HD-02, GSM1808394.HD-03, GSM1808395.HD-04, GSM279060, GSM279061, GSM279062.; B. Density estimates (histograms) for arrays GSM1808377.HT-01, GSM1808378.HT-02, GSM1808379.HT-03, GSM1808380.HT- 04, SM1808381.HT-05, GSM1808382.HT-06, SM1808383.HT-07, GSM1808384.HT-08, GSM1808385.CKD-01, SM1808386.CKD-02, GSM1808387.CKD-03, SM1808388.CKD-04, GSM1808389.CKD-05, GSM1808390.CKD-06, GSM1808391.CKD-07, SM1808392.HD-01, GSM1808393.HD-02, GSM1808394.HD-03, GSM1808395.HD-04, GSM279060, GSM279061, GSM279062; C. NUSE plot.; D. RLE plot.
Intensity plot: Density curves illustrating the distribution of signal intensities in different arrays shown in Figure 2B. Note the peak to the left which corresponds to the background signal: genes expressed below this level will show up within this area. Above this are the probes that are notably expressed. The number of probes decreases exponentially as the intensity increases, not how the curves all have the same, linear slope. However, the upper tail is shifted to the right or left depending on whether the signal is strong or weak.
MA plot: In MA plot, genes with similar expression levels in two experiments will appear around the horizontal line y=0. In our array every chip indicates outliers see Figures 3A1-3A3. An MA-plot is a plot of log-fold change (M-values, that is, the log of the ratio of level counts for each gene between two samples) against the log-average (A-values, that is, the average level counts for each gene across the two samples).

Figure 3: A1, A2 and A3 MA plots: A reference array is calculated from the median across arrays and for each array M and A values are calculated for the comparison to that reference.
NUSE plot: Each box shows NUSE values of the array of Expression files (CEL) that is, NUSEi. In general, value of NUSEi≈1. In our case array i=1,6; i=5,7 and i=4 corresponding to HT, CKD and HD respectively are found deviated from the normal. Thus from the NUSE value one can easily identify lower quality arrays. This is much easier than the Affymetrix quality standards to distinguish problematic arrays (Figure 2C).
QC stats: Each array is represented by a separate line in the Figure 4A. The central vertical line corresponds to fold change, FC=0, the dotted lines on either side correspond to up and down regulation. The blue bar represents the region in which all arrays have scale factors within, by default, three-fold of each other. Its position is found by calculating the mean scale factor for all chips and placing the center of the region such that the borders are -1.5 fold up or down from the mean value. Each array is plotted as a line from the 0-fold line to the point that corresponds to its scale factor. If the ends of all of the lines are in the blue region, their scale-factors are compatible. The lines are colored blue if OK, red if not (Figure 4A).
RNA degradation plot: The degradation plot shows the (shifted and scaled) mean intensity < RNAsi > for each position within probe sets. High slopes indicate degradation. At minimal degradation, we can see an upward trend in expression levels as probe number increase. Therefore,
RNAs1=Avg intensity
Where S represents index for cell files and i the index for probe set ID see in Figure 4B.

Figure 4: A. QC stats.; B. RNA digestion/degradation plots for arrays GSM1808377.HT-01, GSM1808378.HT02, GSM1808379.HT-03, GSM1808380.HT-04, GSM1808381.HT-05, GSM1808382.HT-06, GSM1808383.HT-07, GSM1808384.HT-08, GSM1808385.CKD-01, GSM1808386.CKD-02, GSM1808387.CKD-03, GSM1808388.CKD-04, GSM1808389.CKD-05, GSM1808390.CKD-06, GSM1808391.CKD-07, GSM1808392.HD-01, GSM1808393.HD-02, GSM1808394.HD-03, GSM1808395.HD-04, GSM279060, GSM279061, GSM279062.
RLE plots: The Relative Log Expression (RLE) values were computed by calculating for each probe-set, the ratio between the expression of a probe-set and the median expression of this probe-set across all arrays of the experiment. It is assumed that most probe-sets were not changed across the arrays, so it is expected that these ratios RLE ≅0 on a log scale. The boxplots presenting the distribution of these log-ratios should then be centered near 0 and have similar spread. Other behavior would be a sign of low quality (see in Figure 2D)
Median Absolute Deviation (MAD): Figure 3B shows a false color display of between arrays distances, computed as the MAD of M-values of each pair of arrays.
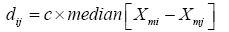
Here, xmi is the normalized intensity value of the mth probe on the ith array, on the original data scale. c=1:4826 is a constant. Pairwise differences between arrays, computed as the MAD of the differences of the M-values.
Differentially Expressed Genes (DEGs) analysis
Total 54675 genes from GSE70528 and 22278 genes from GSE30122 were filtered at FC=6 and 2 respectively. We got 21 Up-regulated genes on comparing the expression of set 1 (HCH and GLOMARULI diabetes genes), while 8 genes were found for the
CKD is the condensed term used for the heterogeneous disorders affecting the kidney structure and function. CKD has formed as the major type of illness worldwide. Cardiovascular diseases are a very frequent implication of CKD nowadays. On the other hand High throughput gene expression recording using microarray has given the opportunity to intelligently curate the fundamental genes. We bolted out and report a set of ten genes that are differentially expressed in various conditions inside kidney. These extracted 10 genes were further divided in to two set. These two set of genes are found actively participating in causing cancer. We hope that the in depth expression and ontological accounts given in this article will pave a new approach to think for the novel therapeutic discoveries.
The author declares that there is no conflict of interest.
This work is financially supported by University Grand Commission (UGC) under NON-NET Fellowship and the Infrastructure (lab facility) was provided by Jamia Millia Islamia.