Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2016) Volume 5, Issue 3
Salmonella enterica is pathogenic bacteria responsible for typhi fever and several gastrointestinal infections. The antibiotics are being used for the treatment of typhi fever and other infections caused by S. enterica. The antibiotic basically target homologous proteins and these bacterial species undergo mutations and develop resistance against particular antibiotics. Also antibiotics have several side effects. Thus there is a need to explore new drug targets to address the issues and side effects of antibiotics. In this paper an attempt has been made to explore riboswitch as a potential drug target for S. enterica. The riboswitches are found in 5’ UTR where mutations are very rare. Also it has been reported that mutations in the riboswitch can be harmful to the organism itself. An in silico approach has been proposed and employed to predict the riboswitches for S. enterica and design of its inhibitor. Seven riboswitches have been predicted using RibEx, out of which one has been taken up in this investigation and structure has been predicted using iFoldRNA. Blind and focused docking has been performed to identify alanine as ligand. Then virtual screening has been performed on the basis of ADMET profiling study and Lipinski’s rule of five against a library of drug like molecules and twelve lead molecules were identified as inhibitors. These inhibitors are having very less side effects. Hence, it can be concluded that riboswitch can be used as alternate drug target for treatment of infectious problems caused by S. enterica and address issues of resistance and side effects of drugs.
<Keywords: Riboswitch; Virtual screening; Autodock; Gastrointestinal; Salmonella enterica
Salmonella enterica is a facultative anaerobic, rod shaped, gramnegative, flagellated bacteria belonging to genus salmonella [1]. Various strains of this bacterium are pathogenic for human being and cause typhi fever via infected food ingested through oral rout. This bacterium affects human and animals but Salmonella enterica typhimurium strain solely affects human being and there is no animal reservoir [2]. The treatment of typhi fever is done with antibiotics like ciprofloxacine [3], chloramphenicol, quinolone and cephalosporin that target the homologous proteins. Some bacterial strains of salmonella have developed resistance to one or more types of antibiotics that is increasingly becoming a problem with typhoid infections arising from south East Asia. The emergence of multidrug resistance to the used antibiotics has complicated the treatment and management of enteric fever [4,5]. So it is must to go for a novel approach that can also target the disease by gene regulation. Research of the past decade has given a novel way of gene regulation at mRNA level which is mediated by Riboswitches [6,7]. Riboswitches are RNA elements that are mainly present on 5’ untranslated region (UTR) of mRNA molecules and able to regulate gene expression in many metabolic pathways. These regulatory elements are consisting of aptamer domain that is conserved and expression platform which is responsible for modulating gene expression as a result of ligand binding [7-10]. The process of drug development is challenging, expensive, and time consuming, although this process has been accelerated due to the development of computational tools and methodologies. The current structure based drug design approach is incomplete because most of the drugs developed by structure guided approaches have been shown to have serious toxic side effects [11-13]. This approach can be applied taking riboswitch as a biological target so that the treatment would be done at gene level and gene regulation will be able to resolve the problem of resistance among various strains.
Tools
NCBI, RibEx [14], Riboswitch Finder [15], Blast, Transcription Translation Tool (www.attotron.com), iFoldRNA [16], AutoDock [17], Raccoon [Stefano Forli, Online], OSIRIS Property Explorer, PharmaGist [18], TEST. The description of various steps followed in the process of designing of inhibitor of designed riboswitch is as follows:
Preparation of input files
The dataset used for study was Salmonella enterica subspecies enterica serovar Typhi Ty2, complete genome having accession number 29140506 was taken from NCBI database. Full genome sequence of Salmonella enterica has been taken from NCBI in FASTA format. This data forms input for the present study.
Identification of gene sequence of riboswitch
The identification is done with the help of RibEx, an online web server. Based on the sequence conservation, RibEx is able to detect specific sequence elements and along with that it is also capable of searching 10 types of riboswitches against an input sequence [16].
Transcription of identified riboswitch
As the homology modelling is possible for RNA sequence, it is needed to convert the obtained DNA sequence into RNA sequence i.e. transcription. The Transcription Translation Tool is employed to transcribe the identified riboswitch like sequence.
Prediction of 3D structure of riboswitch
The secondary and tertiary structure of riboswitch is predicted by using online iFoldRNA tool. iFoldRNA program is a web based methodology for rapidly exploring RNA conformations using discrete molecular dynamics simulations [16]. The 3D structure of the riboswitch is further visualized using GUI interface of the Chimera molecular modeling suite. Chimera is a protractile program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories and conformational ensembles.
Molecular docking simulation
Molecular docking is done to predict the binding affinity between ligand and receptor.
Blind docking: Blind docking is done by using the GUI interface of AutoDock and software is downloaded from the Scripps portal [17-20]. In blind docking; the appropriate binding site is searched in the predicted riboswitch of Salmonella enterica. In the blind docking method, the whole riboswitch structure is covered under 3D grid box for docking. The coordinates of grid box utilized for finding a binding site for the riboswitch as given in Table 1.
| Riboswitch | X-D | Y-D | Z-D | Spacing | X-Center | Y-Center | Z-Center |
|---|---|---|---|---|---|---|---|
| 80 | 72 | 82 | 0.475 | -1.75 | 0.028 | -0.472 |
Table 1: The coordinates of grid box used in blind docking.
As, Alanine is neutral and simplest amino acids among all the 20 amino acids therefore it is used as ligand to predict the binding site in the riboswitch during blind docking. Appropriate binding sites are identified on the basis of lowest binding energy in the range of -5 to -15 Kcal/Mol. The identified binding residue present in the riboswitch is further utilized for focused docking.
Focused docking for ligand identification: For targeting the specific ligand binding site, focused grid box covering ligand as well as binding residues is prepared. The coordinates of grid box used in focused docking of the riboswitch are given in Table 2. Focused docking is performed by using 20 different amino acids and 11 different cofactor or metabolites which are involved in direct binding at 5’UTR of different riboswitches.
| Riboswitch | X-D | Y-D | Z-D | Spacing | X-Center | Y- Center | Z-Center |
|---|---|---|---|---|---|---|---|
| 40 | 40 | 40 | 0.242 | -0.083 | -0.25 | 0.465 |
Table 2: The coordinates of grid box used in focused docking.
Refinement of focused docking results
After identification of an appropriate ligand from focused docking, repeat the docking of identified ligand with the riboswitch number of times to refine the results.
Virtual screening using NCI diversity set
Virtual screening of the NCI diversity set containing 1880 diverse molecules is done by using identified the binding site predicted in the structure of the riboswitch. The files required for virtual screening are prepared through Raccoon software. Raccoon [Stefano Forli, Online] is a graphical interface for processing ligand libraries in different formats (PDB, PDBQT) and to generate all files required to run an AutoDock virtual screening. The coordinates of grid box used in virtual screening of the riboswitch are same as docking and grid parameters used in focused docking as given in Table 2.
Predication of drug score and toxicity of lead molecules
The top five screened lead molecules are checked for toxicity and drug score with the help of OSIRIS online tool. OSIRIS calculate various drug relevant properties like cLogP value, molecular weight, Drug likeness, drug score and toxicities like mutagenicity, tumorigenicity, irritant effects and reproductive effects in lead molecules on the basis of functional group present in the structure of lead molecules. The results predicted in OSIRIS are color coded. Red color shows the properties with high risks of undesired effects like mutagenicity or a poor intestinal absorption, whereas a green color indicates drug-conform behavior. The drug score combines a drug likeness, cLogP, molecular weight and toxicity risk, which is used to judge the lead molecule overall potential to qualify for a drug [OSIRIS Property Explorer, Online]. The predicted results were tested with TEST [Toxicity Estimation Software Tool] software.
The stepwise summary of the results obtained is as follows:
Preparation of input
Gene bifunctional aspartokinase/homoserine dehydrogenase is identified from the genome of Salmonella enterica which contain the riboswitch like element (RLE).
Identification/prediction of gene sequence for riboswitch
After putting the gene sequence into the RibEx online tool, the riboswitch is identified. Sequence of RLE:
CACCACCACCATTACCACCATCACCATTACCACAGGTAAC
Transcription of Identified Riboswitch:
The gene sequence and its transcribed sequence are given below:
Gene sequence
CACCACCACCATTACCACCATCACCATTACCACAGGTAAC
Transcripted sequence
GUGGUGGUGGUAAUGGUGGUAGUGGUAAUGGUGUCCAUUG
Prediction of 3D structure of riboswitch
The tertiary structure of identified from Salmonella enterica as shown in Figure 1, is predicted by iFoldRNA online tool and is visualized by PyMol.

Figure 1: The tertiary structure of RLE.
Docking
Blind and Focused docking is performed.
Blind docking
The whole 3D structure of riboswitch and Alanine is covered under the grid by using coordinate values as given in Table 1 and further docked. Alanine binds to its particular binding site and gives some energy as shown in Table 3.
| Ligand | Binding energy (Kcal/Mol) | Ki (µM) |
|---|---|---|
| Alanine | -7.17 | 740.21 |
Table 3: Blind docking result of riboswitch like element.
Focused docking for ligand identification
The binding sites which we obtained from blind docking is further covered under the grid box by using the coordinates as given in Table 2, and docked with 20 amino acids and 7 metabolites which have an affinity of binding on aptamer site of different riboswitches and are checked for lowest binding energy. The detailed results for focused docking are shown in Table 4. Queosine shows the minimum binding energy of -9.49 Kcal/Mol. Therefore, this ligand was chosen for further refinement process.
| Serial No | Ligand | Binding energy (Kcal/Mol) |
|---|---|---|
| 1. | Alanine | -6.42 |
| 2. | Glycine | -3.59 |
| 3. | Valine | -3.69 |
| 4. | Lysine | -6.46 |
| 5. | Isoleucine | -3.85 |
| 6. | Tyrosine | -5.21 |
| 7. | Tryptophan | -5.16 |
| 8. | Phenylalanine | -4.37 |
| 9. | Proline | -3.77 |
| 10. | Arginine | -7.04 |
| 11. | Histidine | -4.28 |
| 12. | Aspartate | -1.15 |
| 13. | Glutamate | -1.97 |
| 14. | Leucine | -3.96 |
| 15. | Glutamine | -4.58 |
| 16. | Threonine | - |
| 17. | Serine | -3.6 |
| 18. | Methionine | -3.5 |
| 19. | Asparagine | -4.32 |
| 20. | Cysteine | -3.85 |
| 21. | Adenine | -3.97 |
| 22. | d-Glucosamine 6 phosphate | -5.42 |
| 23. | FMN | -2.84 |
| 24. | Guanine | -7.19 |
| 25. | Queosine | -9.49 |
| 26. | s-adenosyl methionine | -4.22 |
| 27. | s-adenosyl thymocysteine | -5.19 |
| 28. | Cyclic di GMP | -2.96 |
Table 4: Focused docking results for riboswitch like element.
Virtual screening using NCI diversity set
Through virtual screening of NCI diversity set containing 1880 diverse molecules, many lead molecules are screened and are arranged according to minimum to maximum binding energy. Among those lead molecules, five lead molecules are selected randomly of different binding energies and are shown in Table 5.
| S.No. | ZINC ID | Structure | Binding Energy |
|---|---|---|---|
| 1 | ZINC01673377 | 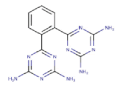 |
-11.55 |
| 2 | ZINC18163421_1 | 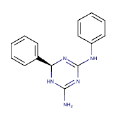 |
-9.93 |
| 3 | ZINC19325794_2 | 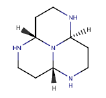 |
-11.23 |
| 4 | ZINC04824902 | 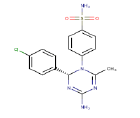 |
-9.6 |
| 5 | ZINC16951318_1 | 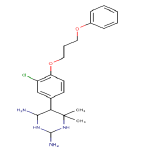 |
-9.22 |
Table 5: Lead molecules with their respective binding energies.
Predication of drug score and toxicity of lead molecules
The toxicity and drug scores of five screened lead molecules predicted by using the OSIRIS online tool for riboswitch are shown in Table 6. Out of five lead molecules, one lead molecule, i.e., ZINC16951318_1 is tumorigenic, have reproductive effect and have a low drug score which means it has a high risk factor. Whereas, other lead molecules like ZINC19362651 and ZINC19325794_2 have no risk factors, that mean they are non-mutagenic, non-irritant, nontumorigenic, do not have any reproductive effect and have a good drug score.
| ID | Mutage nic | Tumorigenic | Irritant | Reproductiv e effect | Drug score | Developmental Toxicant |
|---|---|---|---|---|---|---|
| ZINC01673377 | - | - | + | - | 0.23 | 1.52 (+) |
| ZINC18163421_1 | + | + | + | + | 0.07 | 0.66 (+) |
| ZINC19325794_2 | - | - | - | - | 0.91 | NA |
| ZINC04824902 | NA | NA | NA | NA | NA | NA |
| ZINC19362651 | - | - | - | - | 0.69 | -0.55 (-) |
| ZINC16951318_1 | + | + | + | + | 0.07 | 0.91 (+) |
Table 6: Toxicity and drug score prediction.
Evaluation of physicochemical properties of lead molecules
In physiochemical properties, the Lipinski’s rule of five criteria of four lead molecules, i.e., ZINC01673377, ZINC19325794_2 and ZINC19362651 has been evaluated and the result is given in Table 7.
| Parameter | Optimum range | ZINC0167337 7 | ZINC1932579 4_2 | ZINC1936265 1 |
|---|---|---|---|---|
| ClogP | -5 to +5 | 0.96 | 0.15 | 4.17 |
| PSA(2D) | 60 to140 | 123.3 | 39.33 | 66.87 |
| Mol. Wt. | 150 to 500 | 266 | 182.0 | 342 |
| HBA | <10 | 10 | 8 | 2 |
| HBD | <5 | 14 | 10 | 0 |
Table 7: Physiochemical properties of proposed lead molecules.
The molecule ZINC19325794_2 shows less polar surface area (PSA (2D)) value than the optimum value. The remaining ZINC01673377 and ZINC19362651 lead molecules follow the Lipinski’s rule of five.
On the basis of predicted values, it can be concluded that two lead molecules ZINC01673377 and ZINC19362651 can act as potential inhibitors against the riboswitch of Salmonella species as all lead molecules show good binding energy with the identified bacterial riboswitch with optimum pharmacokinetics properties. These molecules can inhibit growth of Salmonella bacterial species by targeting their riboswitch. Because of the slow metabolism rate of these lead molecules, they will result in longer duration of action in the human body with a smaller therapeutic dose. As riboswitches are conserved throughout the evolutionary process, they can be considered as a better inhibitor against biological target.