Journal of Molecular Imaging & Dynamics
Open Access
ISSN: 2155-9937
ISSN: 2155-9937
Research Article - (2018) Volume 8, Issue 2
Acetylcholinesterase (AChE) is a key enzyme in central nervous system which hydrolaze neurotransmitter acetylcholine (ACh) at cholinergic brain synapses. High expression of AChE in brain and muscles is responsible for diverse neurological conditions leading from neuromuscular, neurodegenerative and neuropsychological diseases. miRNA-132 is a non-coding RNA molecule which help to regulate the expression level of number of genes involved in neurological development, angiogenesis and synaptic transmission. However, miR-132 is the only miR so far that has been experimentally validated as targeting AChE, with consequences on inflammatory responses. Various bioinformatics tools like MODELLER, I-Tasser, ModeRNA, 3D-Dart, ERRAT, ProSA and Rampage were employed for AChE, miR-132 and mRNA model building and their validations. The binding affinities of these macromolecules were also investigated. The results revealed that the miR-132 binds to 3'-UTR region of AChE. The catalytic triad Ser-203, His-447 and Glu-334 found in active site gorge of protein binds to the ligand. Thus, the models of proteinligand and mRNA-miR-132 complexes proved miR-132 as an inhibitor of Acetylcholinesterase. Therefore miR-132 can be a potent inhibitor for Acetylcholinesterase and further analysis of this inhibitor could be helpful for exploration of ligand binding pockets and designing of a novel therapeutic target to cure central nervous system disorders.
Keywords: Acetylcholinesterase; Active site gorge; Huperzine A; miRNA-132; Comparative modeling; Binding affinity
The microRNAs (miRNAs) form a class of small regulating RNA of nearly 22 nucleotides in length. They are able to post-transcriptionally regulate the expression of target mRNAs. In almost all cases, miRNAs interact with the target mRNAs by an imperfect matching occurring between the mRNA 3′-untranslated region i.e. 3′-UTR and a region located between nucleotides 2 and 8 in the 5′ region of the miRNA which is referred to as the miRNA ‘seed’ region. It depends on the complementarity degree of this interaction between the miR and mRNA that miRNA can lead to the cleavage and degradation of their mRNA targets especially when the matching is perfect or nearly perfect or to their translational inhibition that occurs when the interaction involves more mismatches.
Now-a-days over 1500 miRNA-encoding genes have been identified in the human genome, and Bioinformatical analyses based on the degree of complementarity between miRNA “seed” regions and mRNA 3′-UTR. This revelation predicts that dozens to hundreds of mRNAs expression is regulated by any miRNA. In addition to this, a particular gene transcript can be a target of several miRNAs. However, the predicted impact of miRNAs on target mRNAs is probably underestimated since accumulating data indicate that miRNA regions located outside the seed region are involved in mRNA recognition and that miRNAs can bind other regions than mRNA 3′-UTR, including their 5′-UTR and coding sequence. Moreover, miRNAs can also positively regulate gene expression by:
(i) Enhancing translation of mRNA and
(ii) Inducing gene expression by target gene promoter binding, thereby more complexity is added to the initially described mode of action [1,2].
Acetylcholinesterase which is also known as AChE or Acetylhydrolase, is a hydrolase that hydrolyzes the neurotransmitter acetylcholine. The original location of Acetylcholinesterase is at neuromuscular junctions and cholinergic brain synapses, where it serves to terminate synaptic transmission. Acetylcholinesterase belongs to carboxylesterase family of enzyme and is the primary target of inhibition by organophosphorus compounds including nerve agents and pesticides [3]. For over twenty years, Acetylcholinesterase is also involved to play other roles, in addition to its classical role of hydrolyzing Acetylcholine, i.e. in terminating synaptic transmission [4].
The major residues that performs all catalysis are in AChE catalytic site that features a Serine-203, Histadine-47, Glutamate-334 forming a triad is at the bottom of a deep and narrow gorge called as catalytic triad. The inhibition of Acetylcholinesterase can result from binding of inhibitor to either the,
(i) Gorge entrance called the “peripheral anionic site” (PAS) or
(ii) To the active site. The oxyanion hole lies with the active site region of AChE with key residues of Glycine-121, Glycine-122, and Alanine-204.
In active site, also lies another substituent called “acyl pocket” featuring Trypsine-236, Phenylalanine-295, Phenylalanine-297 and Phenylalanine-338.
In silico approach helps in formulating better refined structures in quick time with all information on their atoms and residues. The structures and their active residues can be predicted with limited experimental data by means of software. By using structural bioinformatics domain, the current study is focused on following objectives:
1. The binding affinities of miRNA-132 with mRNA of AChE.
2. To make plausible relationship between these two important regulatory molecules as a repressor or activator.
3. To suggest active residues involved in AChE interacting pathways.
Although many researches have been focused on finding their mechanism of action on various targets, only few researches point out their relation to each other. MicroRNAs add to both roles in cells of nervous and immune system, but their participation in contact between tissues is yet to be explored. It was hypothesized that microRNAs which binds to Acetylcholinesterase (AChE) can assuage inflammation because the brain with secretion of acetylcholine by vagus lessens inflammation by interception of synthesis of cytokine. It was also reported that provocative stimulus induced leukocyte over expression of the miR-132 which binds to Acetylcholinesterase.
The quantity of miR-132 was exhausted by Injected locked nucleic acid (LNA)-modified anti-miR-132 oligonucleotide whilst increasing Acetylcholinesterase quantity in mouse circulation and tissues systems. When cells were transfected by inducing mutation in the 3’-UTR regions of mRNA of AChE when it bonded to miR-132, Acetylcholinesterase expression was elevated. But when the cells were infected by mutation via lentivirus expression system, pre-miR-132 exhibited a declining Acetylcholinesterase expression. Despite of substantial upregulation of miR-132 in brain and bone marrow, transgenic mice overexpress 3’-UTR with no Acetylcholinesterase still showed enhanced mediators of inflammation and non-functional cholinergic anti-inflammatory regulation [5].
Inflammatory constraint is not confined to mere parasympathetic cholinergic signaling. Sympathetic adrenergic, neuroendocrine hypothalamic-pituitary-adrenal (HPA), and hypothalamic-pituitarygonadal (HPG) mechanisms also controls nervous system and immune system [6]. Various neuropeptides present in peripheral nervous system of human body (PNS) also play significant function in calculating immune system. For instance, release of substance P in pancreatic neurons regulates islet inflammation and insulin resistance [7]. Hence, it can be concluded that control of inflammation by miR-132 has potentially represented wide ranged processes.
In communication between body and brain, miR-132 play a role such as the neuro-endocrine intonation of inflammation, in fact it optimizes the inflammation by invoking natural immune system in body with the help of Acetylcholine. In afferent fibers, the vagus nerve signals the occurrence of cytokines in brain which had not caused inflammation. This is called “cholinergic reflex” and it activates inflammatory disease.
Synaptophysin-positive nerve endings at level of tissue, interacts with macrophages of spleen and surgical ablation of the splenic nerve indicating such connections necessary for the cholinergic antiinflammatory management of endotoxemia [8]. Thus, communication of body and brain through Acetylcholine is closely concerned in the control of inflammation. Researchers take mouse and electric fish as a candidate subject for their work regarding Acetylcholinesterase.
Out of nearly 1000 structures deposited in PDB, only few crystal structures of human AChE have been solved but they are recombinant in nature. The reason for this scanty PDB deposits is lack of experimental investigation as extraction of this enzyme involved difficulty and precisions. Huperzine A with its strong affinity as inhibitor to Acetylcholinesterase binds only in few contacting points. First, although Huperzine A has three potential Hydrogen bond donor and acceptor site, only one strong Hydrogen bond has been seen between the pyridine oxygen of the ligand and a protein residue by in silico analysis. Inhibition of human Acetylcholinesterase by tabun agent used during warfare and the succeeding aging response is of picky attention to the scientific community; the conjugate exhibits an unusual position toward nearly all oxime reactivators [5].
Current research methodology stands on the principle of in silico analysis of miR-132 with mRNA of Acetylcholinesterase. Comprehensive computational studies were performed on miR-312 keeping in view its potential role in neurological disorders. A detailed study is mentioned below:
Sequence retrieval and 3D model generation
The amino acid sequence, origin, annotation and functional information of proteins were retrieved from Uniprot Knowledgebase database along with Accession numbers. The template for the construction of miR-132 and mRNA structures was obtained from Rfam by ModeRNA server. miRBase database was analysed to extract the sequence miR-132 in humans.
The sequence of mRNA of Acetylcholinesterase was retrieved by using GenBank which provides sequences of all kinds of RNA of all species. Ligand of target AChE was retrieved from KEGG LIGAND database. NCBI Basic Local Alignment Search Tool was used to locate the best possible template against query protein and mRNA to target homology modeling. The 3D structures of top prioritized templates with query sequence were retrieved from Protein Databank (PDB) [9].
RNAiFOLD is an algorithm for RNA inverse folding and molecular design with constraints programming. The main purpose for using RNAiFOLD was to see how the mRNA structure would be folded and its energy values are also provided by the server for the further 3D modeling of AChE. 3D-Dart server i.e. part of the HADDOCK algorithm was employed for the construction of 3D structure of mRNA with restraints and conditions self-applied [10].
I-Tasser detects structure templates from the Protein Data Bank by a technique called fold recognition or also called as threading. I-TASSER built the 3D structure of Acetylcholinesterase by using LOMETS for multiple threading alignments. In addition to the structure prediction, I-TASSER also performed sequence comparisons, alignments, clustering, structure evaluation, and predicts the de novo modeling of loops. ModeRNA, particular software for RNA 3D structure prediction, analysis and manipulation, was employed for miR-132 and mRNA [11].
Model evaluation
Vast variety of tools like ERRAT, ProSA and Rampage were used to access the errors in the experimentally generated model. Rampage generates a Ramachandran plot to provide basis for the assessment of protein structures in terms of favored, allowed and outlier regions. ERRAT provides user a plot to indicate the confidence and overall quality of the predicted model.
Docking analysis of AChE
Hex webserver was used for AChE-miR-132 docking. For the docking of predicted structure of AChE with suitable ligand, AutoDockVina was applied [12].
Visualization of docked complexes
To visualize and analyze the Protein-Ligand docked complexes and RNA docking, PyMOL software was used. Along with PyMOL, structure of predicted protein structure and RNA complexes were visualized with great precision with Chimera.
Protein interaction analysis
STRING and STITCH databases were applied to investigate the protein-protein interaction derived from different sources. The Human Protein Reference Database (HPRD), Gene Card and BIOGPS servers were employed to provide the detail on gene and its protein.
Protein structural modeling
The sequence of enzyme Acetylcholinesterase was retrieved from the National Center for Biotechnology Information (NCBI) with protein as a database. The selected sequence had the Accession number AAA68151.1. The same protein i.e. AChE can be found in Uniprot Knowledgebase with sequence number P22303.
The NCBI BLAST (Basic Local Alignment Search Tool) database was used to search a suitable template to build a homology model of candidate protein. The best template was selected with Accession number 4EY4 A with maximum coverage of 1098, Query Cover: 88%, E-value 0.0 and 100% Identity [13]. MODELLER was used to give the protein structure in PDB format. A detailed summary on Acetylcholinesterase is shown in tabular form in Table 1.
| Target protein | Acetylcholinesterase |
|---|---|
| Symbol | ACHE |
| Molecular weight | 67376 Daltons |
| Highest occurrence | Early erythroids (73.60%) |
| Major function | AChE activity, cholinesterase activity, beta amyloid binding collagen binding |
| Chromosome location | 7q22 |
| Major transcript | E4-6 |
| Motif | Single Protein 1-31 |
| Domain | Trans Membrane 595-611 |
| Interacting units | 9 |
| Exon count | 8 |
| Post-Translational modification | Asn-296, Asn-495 |
| Isoforms | Isoform T (nucleus), Isoform H (Cell membrane) |
| Most similar gene | BChE (T-score 0.84) |
| Mutations | Mostly Targeted |
| Promotor sites | TBP, Sp1, Eg |
Table 1: Summarized data on acetylcholinesterase from Gene-Card, HPRD and BIOGPS.
I-TASSER was utilized for homology modeling by using Multiple Threading Alignment using 4BDTA as a template and generated five best models in PDB format based on Confidence score or C-score. The best model with the greatest C-score of -0.12, an Estimated TM score of 0.7-0.12 and a calculated RMSD (Root Mean Square Deviation) of 8.0-4.4 Armstong is shown graphically in Figure 1. The model however was not of a good quality based on its score and its template [14].
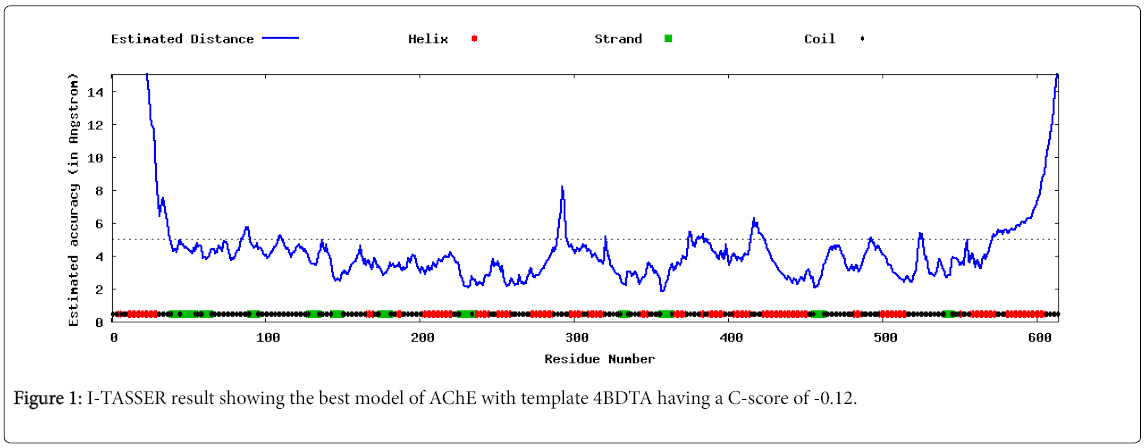
Figure 1: I-TASSER result showing the best model of AChE with template 4BDTA having a C-score of -0.12.
Structural evaluation
The protein model thus obtained was evaluated using RAMPAGE showed as in Figure 2, the protein model as an acceptable structure with 98.0% of residues in favored region and 2.0% in allowed region; while no i.e. 0.0% residues were found in outlier region. The reliability of suspected model was also tested on ERRAT v2.0. The result showed four plots comprising both chain A and chain B with a 92.755% overall quality factor based on 95% to 99% confidence of error function by comparison from highly determined protein structures. ProSA database is utilized to give a Z-score of -10.51 as shown in Figure 2.
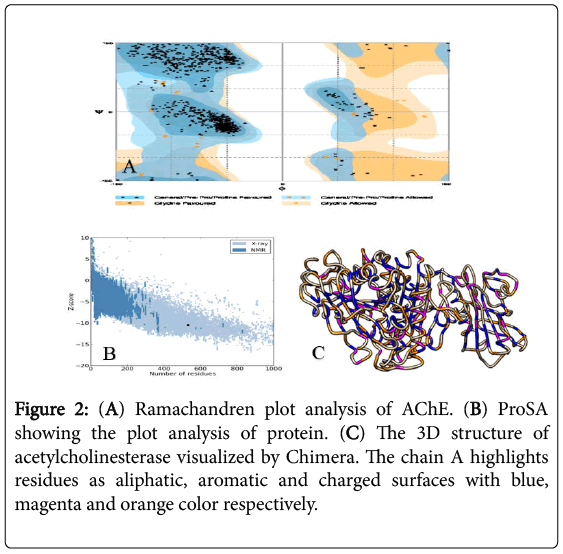
Figure 2: (A) Ramachandren plot analysis of AChE. (B) ProSA showing the plot analysis of protein. (C) The 3D structure of acetylcholinesterase visualized by Chimera. The chain A highlights residues as aliphatic, aromatic and charged surfaces with blue, magenta and orange color respectively.
By using Chimera v.1.10, the 3D structure of Acetylcholinesterase (AChE) was analyzed comprising two chains A and B shown in Figure 2. I-Tasser’s results of the first best model with a C-score of -0.12 are also visualized with Chimera showing the presence of catalytic triad difference of Ser-200, His-440 and Glu-327 because the structure was designed by using Electric fish’s AChE. This is the reason the model from I-Tasser hasn’t been used for docking analysis. The catalytic triad of AChE has been shown in Figure 3.
Figure 3: The 3 catalytic triad of protein visualized by Chimera are close together.
Protein-protein interaction
The databases like STRING (Search Tool for Retrieval of Interacting Genes/Proteins) and STITCH was employed both giving the results for important interacting Proteins with Acetylcholinesterase (Figure 4). The Confidence score of 0.906 from STRING database showed that APP (Amyloid Beta Precursor Protein) although is less than that of COLQ (Collagen like Tail subunit) with highest score 0.966, but the binding and interacting units of AChE with APP has been far better than COLQ evident from the Laboratory analyzed data. STITCH, though used the same principle of query searching and results interpretation as STRING, but all the resulted proteins were inhibitory in action. Rivastigmine showed the highest C-score of 0.998 and can be selected as a candidate for protein-protein interaction [15].
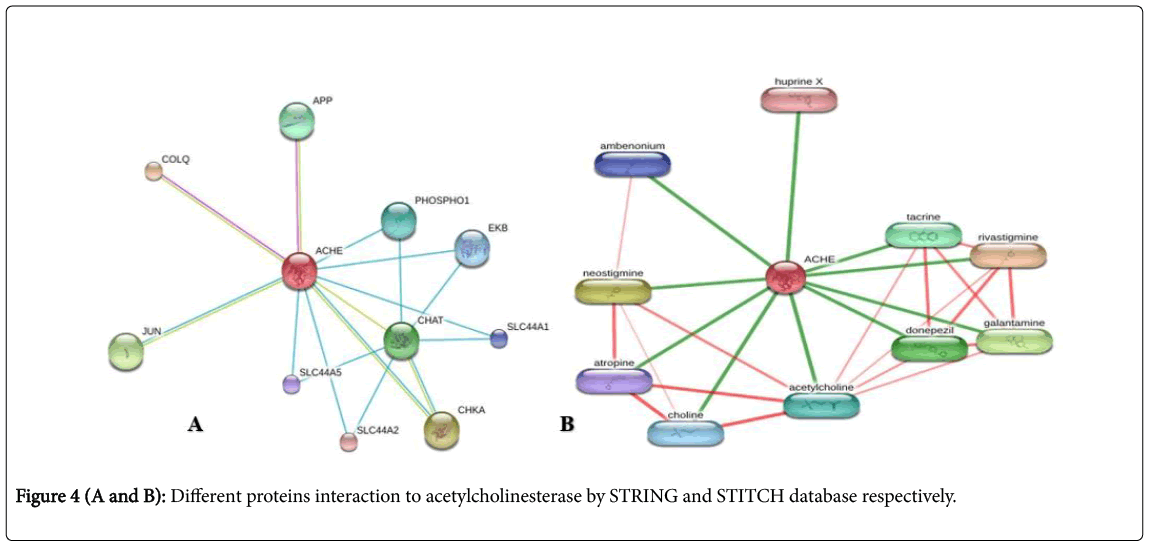
Figure 4(A and B): Different proteins interaction to acetylcholinesterase by STRING and STITCH database respectively.
Ligand retrieval
The ligand Huperzine A was selected from ZINC database (Zinc 19796050) for the protein-ligand docking.
Protein-ligand docking
By keeping the protein and ligand in the same directory, AutoDocVina was run with 1.000 spacing in Angstroms and commands were given for the perfect output having the best conformation and binding mode. Vina generated five results and the best result was the one with the lowest binding affinity i.e. -10.33 kcal/ mol.
The output result was further analyzed by using PyMol as shown in Figure 5. It not only helped in visualizing the docked complex but also gave the knowledge of interacting residues. The major amino acids that’s interacts are the catalytic triad which has been explored in Figure 6. Thus, it proves the binding affinity of ligand Huperzine A with the active site gorge in Acetylcholinesterase [16].

Figure 5: The visualization of cartoon styled acetylcholinesterase bound to ligand in stick by PyMOL.
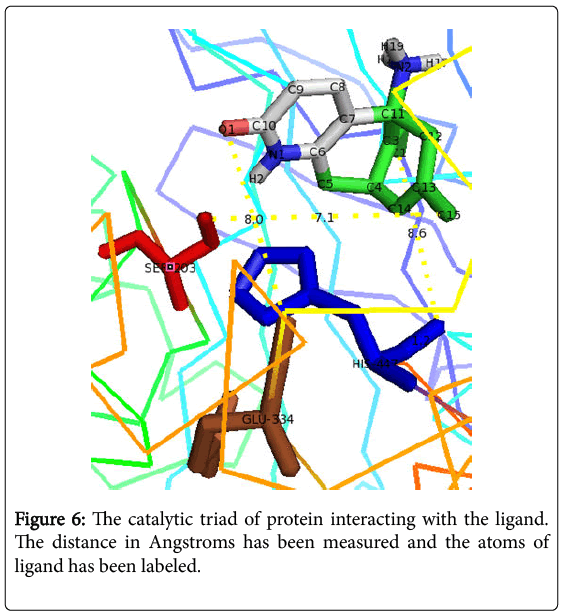
Figure 6: The catalytic triad of protein interacting with the ligand. The distance in Angstroms has been measured and the atoms of ligand has been labeled.
Protein-miR-132 docking
As shown in Figure 7, acetylcholinesterase was first docked with miR-132 to understand the chemistry of Protein-RNA docking. miR-132 docked with protein by using a web-based Hex Server gave the values of -1.00 RMS, -1 Bumps, E total value of -524.15, E-shape of -524.75 and E force was 0.00. The results were visualized using CHIMERA and their interacting residues were highlighted as shown in Figures 8 and 9. The purpose was to analyze the binding affinity of AChE protein with miR-132. miR-132 binds on active site gorge. Further chemistry has been studies by PyMol viewer o understamd residual interaction between two molecules [17].

Figure 7: AChE-miR-132 complex by Hex server.
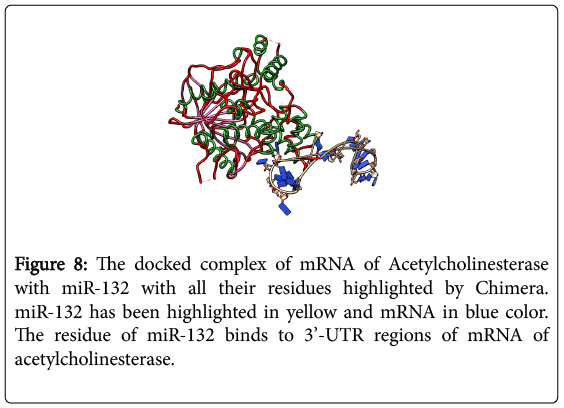
Figure 8: The docked complex of mRNA of Acetylcholinesterase with miR-132 with all their residues highlighted by Chimera. miR-132 has been highlighted in yellow and mRNA in blue color. The residue of miR-132 binds to 3’-UTR regions of mRNA of acetylcholinesterase.

Figure 9: All the atomic interactions of mRNA-miR-132 has been depicted by using PyMOl viewer. mRNA has been shown in colored cartoon style and miR-132 as yellow stick.
The key residues once again in docking with miR-132 were Serine 203, Histidine 447 and Glutamate 334. These three residues make up the catalytic triad that is important in attacking the ligand as validated by research data. Along with catalytic triad, Leu-115, Leu-180, gly-210, Ser-148, Gly-205, Ala-206, Ala-204, Val-183, Gly-121, Trp-117 and Tyr-118 were the nearest residues that interacts with miR-132 and should be the one to form Hydrogen bond with mir-132 [18-20].
miR-132 did not fit correctly into the binding pocket of AChE. As the deduced 3D model of miR-132 is not a perfect model and after the validation by experimental techniques the proper docking mode of miR-132 with protein is yet to be determined, the current pattern of miR-132 and AChE complex only explained the orientation of miR-132 with respect to x, y and z coordinates. The fitting pattern of miR-132 only came half way while the other half structure oriented itself away from the AChE [21].
Considering the way miR-132 interacted with both proteins, it is concluded that miR-132 binds on those regions which lead to decreased activity of AChE as reported in literature survey. The blockage in active site gorge in protein by Huperzine A was done by the catalytic triad in AChE. As surveyed in Literature the active site gorge and the catalytic triad have an inhibitory action on protein when combined with a ligand. First, the protein was docked with Huperzine A, a chemical compound proved the potency of AChE degradation. In the next step, again the protein-miR binding proved that miR-132 also has a potential orientation towards the active site gorge done by Hex server showed the complete binding affinity of both macromolecules, suggesting that miR-132 can naturally cause inhibition of Acetylcholinesterase.